Actu MOC - Avril 2026

Sommaire
🔎 L’achondroplasie, au fil du temps
🔎 Syndrome nail-patella : quand le diagnostic vient du génome non codant
🔎 Ostéogenèse imparfaite : la périostine, un possible marqueur de sévérité
🔎 Setrusumab : quand l’os se densifie… sans moins se fracturer
🔎 FOP : mieux saisir l’expérience des poussées inflammatoires
🔎 Syndrome de Miller : les contours d’une maladie rare se précisent
🔎 Synd. Cornelia de Lange : quand le diagnostic se joue dans plusieurs tissus
L’achondroplasie, au fil du temps
Selon une récente étude internationale, les enfants atteints d’achondroplasie suivent des trajectoires de croissance remarquablement proches d’un pays à l’autre. Mené dans 15 pays, le programme ACHieve apporte l’une des descriptions prospectives les plus complètes à ce jour de l’histoire naturelle de la maladie, et la plus importante cohorte chinoise jamais suivie de façon prospective. De quoi mieux comprendre ce qui, dans l’achondroplasie, relève d’un parcours commun… et ce qui continue de peser sur la santé des enfants.
Pendant longtemps, la connaissance de l’achondroplasie a reposé surtout sur des séries rétrospectives, souvent limitées à quelques pays. Or cette maladie génétique, liée à des variants de FGFR3, ne se résume pas à une petite taille. Elle s’accompagne aussi d’atteintes multiples, touchant la croissance, les proportions corporelles, la respiration, la sphère ORL, la colonne vertébrale ou encore le foramen magnum (orifice de communication entre la cavité crânienne et le canal vertébral). Pour mieux décrire cette évolution dans la « vraie vie », les chercheurs ont suivi 259 enfants âgés de 0 à 8 ans, sans traitement stimulant la croissance, avec des évaluations répétées tous les six mois. Le suivi médian était de 21 mois.
Cette étude rapporte la grande cohérence des trajectoires de croissance entre les régions du monde. La vitesse de croissance diminue rapidement pendant les premières années de vie, puis ralentit plus progressivement jusqu’à environ 4 ans. À 1 an, elle était en moyenne de 9,3 cm/an chez les garçons et 10,4 cm/an chez les filles ; à 4 ans, elle était de 4,1 cm/an et 4,6 cm/an, respectivement. Cette évolution était globalement similaire chez les garçons et les filles, ainsi qu’entre la Chine et les autres régions étudiées. De même, les proportions corporelles, notamment le rapport entre segment supérieur et segment inférieur, restaient globalement comparables d’un pays à l’autre. Ainsi, malgré les différences de contexte médical ou de diagnostic, l’achondroplasie semble suivre un schéma de croissance largement partagé.
Une histoire naturelle, mais pas sans complications
Au cours du suivi, 77,2 % des participants ont présenté au moins un événement clinique, et plus d’un tiers de ces événements ont été considérés comme liés à la maladie. Les plus fréquents étaient les infections respiratoires hautes, la fièvre et les rhinopharyngites, tandis que parmi les événements sérieux figuraient notamment les pneumonies, la sténose du foramen magnum, le syndrome d’apnées obstructives du sommeil, les épanchements de l’oreille moyenne et la sténose cervicale.
L’étude met aussi en lumière la différence dans l’âge du diagnostic selon les régions. En Chine, il survenait en médiane vers 52 semaines, contre environ 2 semaines ailleurs. Pourtant, une fois les enfants inclus dans l’étude, leurs paramètres de croissance apparaissaient très proches. Ce décalage suggère que les différences portent moins sur la biologie de la maladie que sur sa détection précoce et l’organisation des parcours de soins.
On l’aura compris, ce travail ne propose pas un nouveau traitement. Mais il fournit quelque chose de tout aussi important : une cartographie robuste de l’évolution spontanée de l’achondroplasie chez l’enfant. Une base essentielle pour mieux accompagner les familles, repérer les complications, et évaluer plus justement l’effet des nouvelles approches thérapeutiques. Avant de modifier l’histoire naturelle d’une maladie, encore faut-il bien la connaître.
Pour en savoir + : Longitudinal Observation of Children with Achondroplasia: Findings from a Global Natural History Study (ACHieve).
McDonnell C, Hove HB, Irving M, White KK, Fontecha CG, Legare JM, Högler W, Hoernschemeyer DG, Schnabel D, Unger S, Bacino CA, Hofman P, Yu Y, Ma H, Gong C, Luo X, Burrow TA, Baujat G, Mora S, Fiscaletti M, Zhao C, Makara MA, Shu AD, Savarirayan R.Horm Res Paediatr. 2026 Jan 8:1-12. doi: 10.1159/000550169.
Syndrome nail-patella : quand le diagnostic vient du génome non codant
Le syndrome nail-patella est une maladie rare connue pour associer des atteintes squelettiques, rénales et oculaires, avec une expression très variable d’un patient à l’autre. Dans la majorité des cas, le diagnostic moléculaire repose sur l’identification d’un variant pathogène dans LMX1B. Mais certains patients ayant des symptômes très évocateurs restaient sans explication génétique. Une étude menée par les centres de référence des maladies osseuses constitutionnelles montre que l’explication peut se situer ailleurs : dans les régions régulatrices de l’ADN, ces séquences qui contrôlent l’expression des gènes, qu’elles peuvent « éteindre » ou « allumer », comme des interrupteurs.
Les auteurs décrivent quatre familles porteuses d’anomalies génétiques différentes : une délétion de séquences régulatrices, deux remaniements chromosomiques empêchant le dialogue normal entre ces régions régulatrices et le gène, et un variant situé dans la région 5’UTR, modifiant la lecture du message génétique. Tous ces mécanismes aboutissent à une même conséquence : LMX1B n’est plus exprimé correctement, mais avec des répercussions variables selon les tissus concernés.
L’un des apports majeurs de l’étude est de montrer que toutes les anomalies de LMX1B n’ont pas les mêmes conséquences cliniques. Lorsque l’atteinte concerne seulement des éléments régulateurs impliqués dans le développement des membres, le tableau peut rester essentiellement squelettique, sans atteinte rénale ni oculaire identifiée à ce stade. À l’inverse, une anomalie perturbant plus largement l’expression du gène expose davantage au tableau classique du syndrome nail-patella. Autrement dit, la nature précise de l’anomalie moléculaire pourrait aider à adapter la surveillance clinique, notamment sur les plans rénal et ophtalmologique.
Cette étude illustre aussi une évolution importante du diagnostic génétique : il ne suffit plus de rechercher des anomalies dans les gènes eux-mêmes. Il faut aussi explorer les régions non codantes qui en contrôlent l’activité. En intégrant ces mécanismes régulateurs et les remaniements structuraux, les auteurs estiment que le rendement diagnostique du syndrome nail-patella approche désormais 100 % dans leur expérience. Une avancée qui rappelle qu’en génétique, la maladie peut parfois se loger non dans le gène, mais dans ce qui règle son fonctionnement !
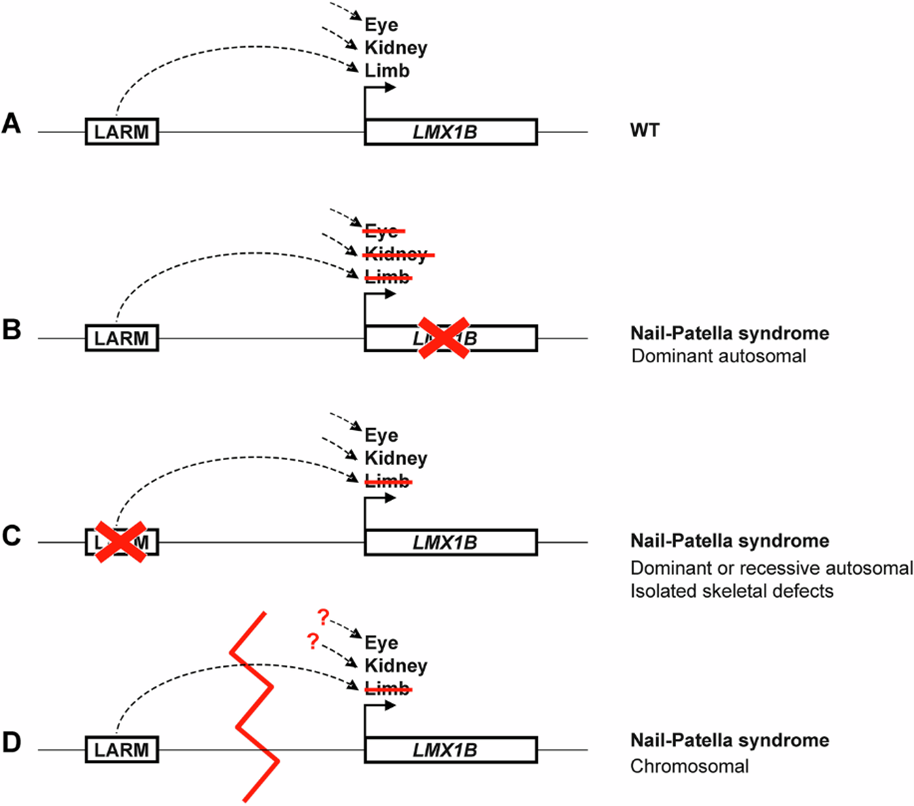
Résumé des mécanismes moléculaires impliqués dans le syndrome nail-patella.
A. Locus de type sauvage (WT, wild type), non représenté à l’échelle.
B. Forme typique du syndrome nail-patella liée à une anomalie du gène.
C. Forme du syndrome nail-patella limitée au squelette, liée à une anomalie des modules autorégulateurs de LMX1B spécifiques du membre (LARM, LMX1B Limb AutoRegulatory Modules).
D. Syndrome nail-patella dû à une variation structurale du génome (ligne rouge discontinue) perturbant l’interaction entre l’enhancer et le promoteur (ligne pointillée).
Résumé des mécanismes moléculaires impliqués dans le syndrome nail-patella.
A. Locus de type sauvage (WT, wild type), non représenté à l’échelle.
B. Forme typique du syndrome nail-patella liée à une anomalie du gène.
C. Forme du syndrome nail-patella limitée au squelette, liée à une anomalie des modules autorégulateurs de LMX1B spécifiques du membre (LARM, LMX1B Limb AutoRegulatory Modules).
D. Syndrome nail-patella dû à une variation structurale du génome (ligne rouge discontinue) perturbant l’interaction entre l’enhancer et le promoteur (ligne pointillée).
Pour en savoir + : Non-coding genome in nail-patella syndrome: Genetic diagnosis as a guide for personalized follow-up.
Brunelle P, Jourdain AS, Escande F, Audebert-Bellanger S, Bouquillon S, Cormier-Daire V, Desdoits A, Lacombe D, Molin A, Gruchy N, Touraine R, Van Gils J, Ghoumid J, Thomes L, Petit F.
Ostéogenèse imparfaite : la périostine, un possible marqueur de sévérité
Pendant longtemps, l’ostéogenèse imparfaite (OI) a surtout été abordée à travers ses manifestations les plus visibles : fractures répétées, déformations osseuses, scoliose, petite taille… Mais au-delà de ces signes, les cliniciens manquent encore d’outils biologiques simples pour apprécier la sévérité de la maladie chez l’adulte. C’est dans cette perspective qu’une équipe lyonnaise s’est intéressée à la périostine, une protéine de la matrice extracellulaire impliquée dans la formation osseuse, le remodelage et la réponse aux contraintes mécaniques.
L’idée n’est pas venue de nulle part. La périostine interagit avec le collagène de type I, justement au cœur de l’OI, et participe à plusieurs voies de signalisation importantes pour le tissu osseux. Les chercheurs ont donc voulu savoir si cette protéine, mesurable dans le sang, pouvait refléter certains aspects de la maladie. Pour cela, ils ont comparé ses concentrations sériques chez 61 adultes atteints d’OI et 61 témoins appariés sur l’âge, le sexe et l’indice de masse corporelle.
Les taux de périostine étaient plus élevés chez les patients OI. Au sein même de la cohorte, les auteurs ont également observé des concentrations plus élevées chez les femmes, chez les patients présentant une scoliose, et chez ceux atteints d’une forme sévère de type III selon la classification de Sillence. Mais sur ce dernier point, ils appellent à la prudence : la cohorte comprenait surtout des formes de type I, et très peu de patients de type III.
Les analyses ont aussi mis en évidence que la périostine semblait liée à plusieurs signes de sévérité de la maladie, notamment certaines atteintes de la colonne vertébrale. Mais après analyses statistiques approfondies, un seul lien restait vraiment solide : les patients ayant eu le plus de fractures sévères présentaient aussi les taux de périostine les plus élevés. À l’inverse, le tabagisme était associé à des taux plus bas (probablement parce qu’il altère le remodelage osseux et la production de matrice extracellulaire).
Cette étude ne fait pas encore de la périostine un biomarqueur prêt à entrer dans la pratique courante. En effet, l’effectif reste limité, il n’existe pas encore de cohorte indépendante de validation, et la protéine n’est pas spécifique de l’os puisqu’elle est aussi exprimée dans d’autres tissus. Mais elle ouvre une piste car pour la première fois, elle suggère qu’un marqueur circulant pourrait aider à mieux saisir, chez l’adulte, la sévérité de l’OI.
Pour en savoir + : Elevated periostin level in serum of adults with Osteogenesis Imperfecta is associated with disease severity.
Mercier-Guery A, Auroux M, Gineyts E, Borel O, Szulc P, Sornay-Rendu E, Fontanges E, Croset M, Rousseau JC, Chapurlat R.J Clin Endocrinol Metab. 2026 Mar 17:dgag119. doi: 10.1210/clinem/dgag119.
Setrusumab : quand l’os se densifie… sans moins se fracturer
Les essais de phase III ORBIT et COSMIC du setrusumab, développés dans l’ostéogenèse imparfaite (OI), livrent un résultat déroutant : la densité minérale osseuse (DMO) augmente nettement, mais sans démonstration statistiquement significative d’une réduction des fractures. Un paradoxe qui oblige à reposer une question centrale : en OI, comment mesurer vraiment l’efficacité d’un traitement ?
Dans ORBIT, mené chez des patients de 5 à 25 ans contre placebo, comme dans COSMIC, conduit chez des enfants de 2 à moins de 7 ans contre bisphosphonates intraveineux, le setrusumab n’a pas atteint son critère principal, fondé sur la réduction du taux annualisé de fractures cliniques.
Les deux études ont en revanche montré un gain significatif de densité minérale osseuse (DMO), sans modification du profil de sécurité. Ces résultats rappellent une réalité souvent sous-estimée : en OI, densifier l’os ne suffit peut-être pas à prouver qu’on le rend moins « cassant ». La discordance entre la DMO et la survenue de fractures affaiblit la pertinence d’un critère substitutif simple et souligne l’intérêt d’endpoints plus directement cliniques, reposant non seulement sur des fractures confirmées radiologiquement, mais aussi sur la douleur, la fonction, l’activité physique et la qualité de vie.
Des analyses complémentaires ont toutefois mis en avant des signaux exploratoires sur les fractures vertébrales et certains « outcomes » rapportés par les patients concernant la douleur et les activités quotidiennes chez les enfants et les adolescents. La société indiquait encore que des discussions réglementaires restaient envisagées.
FOP : mieux saisir l’expérience des poussées inflammatoires
Dans la fibrodysplasie ossifiante progressive (FOP), les poussées inflammatoires constituent des épisodes cliniques majeurs, mais leur vécu restait encore insuffisamment documenté du point de vue des patients. Cette étude qualitative valide le contenu du Flares Diary, un outil de recueil des symptômes de poussée, en montrant qu’il couvre des manifestations jugées pertinentes, compréhensibles et représentatives par des adultes atteints de FOP.
La FOP est une maladie très rare caractérisée par une ossification hétérotopique progressive des tissus conjonctifs. Les poussées, ou flare-ups, s’accompagnent classiquement de gonflement, douleur, raideur, chaleur locale et diminution des mouvements, avec un retentissement important sur la vie quotidienne. Pour évaluer la validité de contenu du Flares Diary, les auteurs ont conduit des entretiens qualitatifs semi-structurés auprès de 20 adultes atteints de FOP, recrutés soit dans l’essai de phase 2 LUMINA-1, soit via l’association internationale des patients. L’objectif n’était pas de mesurer l’efficacité d’un traitement, mais de vérifier si l’outil décrivait correctement l’expérience vécue des poussées.
L’analyse thématique des entretiens a permis d’identifier un ensemble cohérent de symptômes locaux et systémiques. Les plus fréquemment rapportés étaient la croissance osseuse, le gonflement, la douleur, la raideur articulaire, la diminution des mouvements, ainsi que la chaleur et la rougeur. Certains participants ont aussi décrit des manifestations plus générales, comme une sensation de chaud ou froid diffus, des vertiges, de la fièvre ou une modification de l’appétit.
Au-delà des symptômes, l’étude met aussi en lumière le poids fonctionnel et émotionnel des poussées. Les participants décrivent des limitations dans les activités de la vie quotidienne, la mobilité, les tâches instrumentales, le sommeil, mais aussi l’anxiété liée à l’imprévisibilité des épisodes et à la crainte d’une perte fonctionnelle supplémentaire. Les auteurs soulignent ainsi que les poussées ne se résument pas à un événement inflammatoire local, mais constituent une expérience globale, physique et psychologique.
Concernant l’outil lui-même, les retours sont globalement favorables. Les participants ont jugé les items du Flares Diary pertinents et faciles à comprendre, avec peu de difficultés majeures. Quelques nuances apparaissent néanmoins : certains ont du mal à distinguer une nouvelle poussée d’une poussée en cours, ou à savoir avec certitude si une poussée a abouti à une nouvelle ossification hétérotopique sans imagerie. D’autres estiment que certaines notions, comme la raideur et la diminution des mouvements, se recoupent partiellement. Malgré cela, les auteurs concluent que l’outil est adapté pour documenter les poussées dans les essais cliniques futurs.
Pour en savoir + : Content Validation of the Flares Diary: A Qualitative Analysis of the Flares Experience Within the Fibrodysplasia Ossificans Progressiva (FOP) Population.
Baldasaro J, Sanchez RJ, Hartford C, Dahir KM, Keen R, Funck-Brentano T, Pignolo RJ, Davis M, Altman DE.Adv Ther. 2026 Mar 10. doi: 10.1007/s12325-026-03506-6.
Syndrome de Miller : les contours d’une maladie rare se précisent
À partir de la plus grande cohorte publiée à ce jour, une équipe française précise les contours cliniques du syndrome de Miller, une acrofaciodysostose rare liée à des variants bialléliques de DHODH. L’étude rassemble 10 patients issus de 7 familles et confirme les signes cardinaux de la maladie, tout en faisant émerger plusieurs manifestations jusque-là peu décrites.
Le syndrome reste exceptionnellement rare et son phénotype encore imparfaitement balisé. Dans cette série multicentrique, les auteurs montrent que les anomalies postaxiales des membres notamment l’absence du 5e doigt ou orteil demeurent un marqueur majeur. Mais elles s’accompagnent souvent d’une atteinte préaxiale, avec hypoplasie du pouce ou du tibia, ce qui élargit la lecture classique du tableau. La camptodactylie apparaît également plus fréquente qu’attendu. Les figurants illustrent bien cette atteinte des segments acroméliques et mésoméliques, avec synostoses radio-ulnaires, syndactylies et anomalies des rayons.
L’étude apporte aussi plusieurs signaux nouveaux. Des cardiopathies congénitales, principalement des communications interauriculaires, sont retrouvées chez plusieurs patients. Les auteurs décrivent également une atrophie optique dans une famille consanguine, ainsi qu’un nævus simplex facial chez deux enfants, deux observations qui viennent enrichir le spectre phénotypique. À l’inverse, tous les patients vivants de la cohorte présentent un neurodéveloppement normal, ce qui tempère certaines descriptions antérieures.
Sur le plan diagnostique, ce travail plaide pour une évocation plus précoce du syndrome de Miller, y compris en période prénatale, devant l’association d’anomalies des membres, de synostoses et de malformations cardiaques. Les auteurs rappellent qu’une telle reconnaissance peut orienter vers un séquençage de DHODH, faciliter le conseil génétique et organiser une prise en charge multidisciplinaire, avec notamment des évaluations cardiaque, ophtalmologique et auditive.
Pour en savoir + : The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
Aubert Mucca M, Brunelle P, Doco Fenzy M, Vanlerberghe C, Dieux A, Feyereisen L, Jobic F, Lode L, Le Guyader G, Petit F, Schaefer E, Zaafrane-Khachnaoui K, Ziegler A, Patat O.Clin Genet. 2026 Jan;109(1):188-193. doi: 10.1111/cge.70015. Epub 2025 Jul 17.
Synd. Cornelia de Lange : quand le diagnostic se joue dans plusieurs tissus
Le syndrome de Cornelia de Lange (CdLS) fait partie de ces maladies dont le visage clinique est souvent reconnaissable, mais dont le diagnostic moléculaire peut rester étonnamment difficile à établir. Dysmorphie faciale caractéristique, malformations des membres supérieurs, retard de croissance sévère avant et après la naissance, déficience intellectuelle… à ces signes s’ajoutent fréquemment des atteintes digestives, cardiovasculaires, neurologiques, visuelles ou auditives. Pourtant, malgré cette forte cohérence clinique, tous les patients ne trouvent pas immédiatement d’explication génétique.
Dans cette série de 224 patients explorés à l’hôpital Necker sur vingt ans, une anomalie génétique a été identifiée chez 155 d’entre eux, le plus souvent dans NIPBL, principal gène impliqué dans la maladie. Mais, l’un des points majeurs de ce travail est l’importance du mosaïcisme somatique. Ce terme désigne une situation dans laquelle la mutation n’est pas présente dans toutes les cellules de l’organisme, mais seulement dans une partie d’entre elles. Concrètement, cela signifie qu’un variant peut être retrouvé dans la salive ou la peau, tout en restant absent du sang.
Chez plusieurs patients sans anomalie retrouvée d’emblée, l’étude du profil de méthylation de l’ADN, puis l’analyse du génome entier sur plusieurs tissus, ont permis d’identifier des variants introniques profonds de NIPBL, invisibles par les approches classiques. L’étude de l’ARN a confirmé, dans plusieurs cas, leur effet sur l’épissage. Ainsi, dans le syndrome de Cornelia de Lange, le diagnostic ne repose pas toujours sur une seule analyse sanguine. Il faut parfois croiser plusieurs tissus et plusieurs niveaux d’analyse (séquençage, méthylation, ARN) pour faire émerger l’anomalie en cause et améliorer le rendement diagnostique.
Pour en savoir + : P036 Intérêt d’une approche multi-omiques et multi-tissus dans le diagnostic du syndrome de Cornelia de Lange.
Sophie RONDEAU (Paris), Céline HUBER, Ghislaine ROYER, Anne-Laure TOURRE, Marcia HENRY, Bekim SADIKOVIC, Valerie CORMIER-DAIRE / https://assises-genetique.org/sessions-de-posters-presentes/


