Actu CAP - Avril 2026

Sommaire
🔎 Pathologies rares du métabolisme phospho-calcique : le génome affine le diagnostic
🔎 Petite taille de l’enfant : vers un usage plus ciblé des tests génétiques
🔎 XLH : quel effet du traitement conventionnel sur les tissus dentoalvéolaires ?
🔎 XLH : que se passe-t-il quand le burosumab s’arrête à la fin de la croissance ?
🔎 Hypophosphatasie : quand la biologie parle avant les symptômes
🔎 Hypophosphatasie : des résultats de phase III contrastés pour l’efzimfotase alfa
🔎 Hypoparathyroïdie chronique : sortir du prisme de la seule hypocalcémie
🔎 L’hypoparathyroïdie : quand l’os retrouve son rythme ?
Pathologies rares du métabolisme phospho-calcique : le génome affine le diagnostic
Cette communication présentée aux 13ème assises de génétique humaine et médicale dresse un état des lieux de la préindication « Pathologies rares du métabolisme phospho-calcique » à partir des génomes analysés dans le cadre du Plan France Médecine Génomique 2025.
Entre 2020 et septembre 2025, 72 dossiers ont été rendus par SeqOIA et 37 par Auragen. Les situations cliniques couvertes étaient variées : hyper- et hypoparathyroïdies, hypophosphatémies, hypercalcémies infantiles, anomalies de l’amélogenèse, néphrocalcinoses, rachitismes vitamino-résistants et formes syndromiques.
Le rendement diagnostique global atteignait 39 % pour Auragen contre 18 % pour SeqOIA. Cet écart s’explique en partie par le fait qu’Auragen a comptabilisé comme « positifs » les cas porteurs hétérozygotes de SLC34A1 et SLC34A3, non pas comme des causes mendéliennes strictes, mais comme des facteurs de susceptibilité reconnus (PMID: 39461557). En excluant ces cas, le rendement diagnostique d’Auragen diminue à 28 %.
Les performances variaient selon les indications, avec de meilleurs résultats dans les hypophosphatémies avec rachitisme, pour lesquelles 11 diagnostics ont été posés, et des rendements plus faibles dans les hyper- et hypoparathyroïdies. Le gène PHEX impliqué dans le rachitisme hypophosphatémique, était le plus souvent retrouvé. L’un des messages forts de ce travail est que le séquençage du génome permet d’aller au-delà des panels ciblés habituels. Il a notamment permis d’identifier des variants introniques profonds, des remaniements structuraux complexes, mais aussi des gènes inattendus, ce qui élargit le champ diagnostique. Par exemple, un cas de cholestase héréditaire liée à SLC51A a été diagnostiqué à partir d’une carence en vitamine D, permettant une prise en charge adaptée. Un autre cas a révélé un syndrome de Rubinstein-Taybi lié à EP300, associé à une hypoparathyroïdie.
Les autres gènes retrouvés dans les cas positifs relevaient du spectre classique de ces pathologies : AIRE, CASR, ORAI1, ALPL, MMP20, GNA11, STX16, CDC73, SLC34A1, SLC34A3, WNT10A, ENPP1 et DLX3. Par ailleurs, 7 dossiers au total, soit 6,5 %, ont été rendus avec un variant de signification indéterminée.
Cette étude montre que le génome peut apporter des diagnostics supplémentaires par rapport aux approches ciblées, en particulier dans les rachitismes hypophosphatémiques, grâce à une meilleure détection des anomalies complexes et à l’identification de causes inattendues. En revanche, certaines indications, comme les hypo- et hyperparathyroïdies, gardent un rendement plus faible, probablement parce que le génome intervient souvent après un premier bilan génétique déjà négatif. Ce travail a été rendu possible grâce aux données du Plan France Médecine Génomique 2025.
Pour en savoir + : P712 - Pathologies rares du métabolisme phospho-calcique : bilan de la préindication en 2026 (laboratoires Seqoia et Auragen). Louis LEBRETON, Audrey BRIAND-SULEAU, Victor MARIN, Marine SERVEAUX-DANCER, Maureen LOPEZ, Consortium AURAGEN, Arnaud MOLIN, Nicolas RICHARD, Céline GAUCHER, Jérôme BOULIGANT, Laurence PACOT / https://assises-genetique.org/sessions-de-posters-presentes/
Petite taille de l’enfant : vers un usage plus ciblé des tests génétiques
Une recommandation internationale publiée en 2026 propose un cadre structuré pour le recours aux tests génétiques chez les enfants présentant une petite taille inexpliquée. Cette recommandation redéfinit la démarche diagnostique en accordant une place centrale à la recherche de causes génétiques rares. Elle souligne que, derrière un symptôme fréquent en pédiatrie, peuvent se cacher des maladies rares dont l’identification peut modifier la prise en charge, la surveillance et le conseil génétique.
Tous les enfants de petite taille ne relèvent pas d’une exploration génétique systématique, mais certains profils cliniques augmentent nettement la probabilité d’une étiologie rare. Après une évaluation initiale complète, les auteurs proposent de distinguer une petite taille isolée et une petite taille non isolée, cette dernière pouvant relever notamment des enfants présentant une dysplasie squelettique suspectée, une petite taille syndromique avec dysmorphie, malformations ou troubles neuro-développementaux, une atteinte génétique de l’axe GH/IGF-1, une naissance petite pour l’âge gestationnel avec suspicion de trouble de l’empreinte, ou encore une petite taille sévère, disproportionnée, associée à une microcéphalie ou à une macrocéphalie relative. Dans ces situations, la génétique devient un outil majeur pour mettre au jour une maladie rare jusque-là non reconnue.
La recommandation s’appuie sur une revue systématique montrant que le rendement diagnostique varie fortement selon le phénotype. Il est relativement limité dans les petites tailles dites idiopathiques, autour de 15,1 %, mais beaucoup plus élevé dans les formes syndromiques (50,8 %) et surtout dans les situations évocatrices de dysplasie squelettique (69,8 %). Ces données confirment que la génétique est particulièrement utile lorsqu’il existe des éléments en faveur d’une maladie rare osseuse ou syndromique.
Le texte propose ainsi un algorithme pratique, résumé dans la figure ci-dessous, qui distingue plusieurs grands cadres diagnostiques : petite taille isolée, dysplasie squelettique, défaut de l’axe GH/IGF, et forme syndromique. Parmi les diagnostics rares cités ou directement concernés figurent notamment les dysplasies squelettiques génétiques, le syndrome de Turner, les troubles de l’empreinte comme les syndromes de Silver-Russell et de Temple, certaines formes de déficit congénital hypophysaire, ainsi que des anomalies de gènes du cartilage ou de la plaque de croissance comme SHOX, ACAN, NPR2, FGFR3, COL2A1 ou IHH.
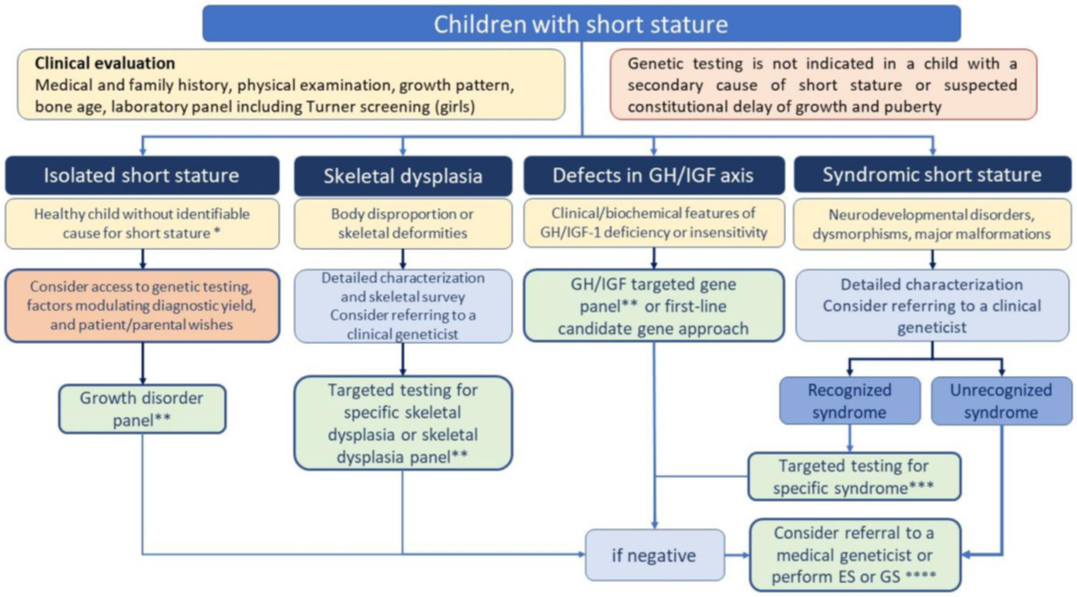
Pour en savoir + : International guideline on genetic testing of children with short stature
Dauber A, Jorge AAL, Nilsson O, Dekkers OM, Argente J, Netchine I, Backeljauw P, Baron J, Bertola DR, Clayton P, Davies JH, Edouard T, Eggermann T, Gevers EF, Grigelioniene G, Heath KE, Jee YH, Lapunzina P, Mortier GR, Pruhova S, Storr HL, Wakeling E, Ferreira CR, Hasegawa T, Hokken-Koelega ACS, Linglart A, Luo X, Wang X, Hwa V, Gregory LC, Buonocore F, Dattani MT, Cianfarani S, Wit JM.Eur J Endocrinol. 2026 Feb 4;194(2):R17-R36. doi: 10.1093/ejendo/lvag013.
XLH : quel effet du traitement conventionnel sur les tissus dentoalvéolaires ?
Dans l’hypophosphatémie liée à l’X (XLH), le traitement conventionnel par suppléments oraux de phosphate et vitamine D active améliore le phénotype osseux, mais son impact sur les tissus dentoalvéolaires restait insuffisamment documenté. Cette étude montre qu’il ne corrige pas significativement les anomalies dentaires et parodontales chez la souris Hyp traitée à partir du stade prépubertaire, tout en suggérant, à partir d’échantillons dentaires humains, un effet favorable sur la qualité de la dentine.
Les auteurs ont combiné deux approches. Chez la souris Hyp, modèle murin de XLH, un traitement associant phosphate oral et calcitriol a été administré de 3 semaines à 3 mois, puis évalué par micro-CT et histologie. En parallèle, des analyses histologiques ont été réalisées sur des dents de patients atteints de XLH traités pendant l’enfance, comparées à des dents témoins.
Chez la souris, les résultats sont globalement négatifs pour la sphère dentoalvéolaire. En effet, le traitement ne restaure ni le volume ni la densité dentine/cément, ne réduit pas l’élargissement des chambres pulpaires, n’améliore pas significativement les paramètres de l’os alvéolaire, et ne corrige pas les défauts d’attache parodontale ni la dentinomalacie.
L’analyse des dents humaines apporte cependant un signal plus nuancé. Les auteurs observent la persistance d’une prédentine augmentée et d’anomalies de minéralisation dentinaire chez les patients traités, mais aussi une amélioration qualitative de la dentine, avec une couche de dentine globulaire anormale paraissant plus limitée chez les patients ayant une meilleure observance thérapeutique. L’interprétation reste prudente, notamment en raison du faible nombre d’échantillons et de leur hétérogénéité, mais ces observations soutiennent l’idée d’un bénéfice partiel du traitement sur la dentine humaine.
Comment expliquer ce décalage entre souris et dents humaines ? Les auteurs avancent plusieurs hypothèses. Chez la souris, le traitement débute à un moment où une partie importante de la dentinogenèse est déjà commencée. Ils rappellent aussi que les tissus dentaires ont une capacité de régénération limitée, et que l’effet du traitement dépend probablement de la précocité d’instauration, de la durée d’exposition et de l’observance thérapeutique.
Ainsi, le traitement conventionnel ne semble pas suffire à corriger pleinement les atteintes dentoalvéolaires de la XLH, même s’il pourrait contribuer à améliorer la minéralisation dentinaire et, possiblement, à réduire le risque d’abcès dentaires spontanés. L’étude souligne ainsi l’importance d’une surveillance bucco-dentaire prolongée, en complément de la prise en charge osseuse.
Pour en savoir + : Impact of oral phosphate supplements and active vitamin D treatment on dentoalveolar features of X-linked hypophosphatemia.
Po J, Lira Dos Santos EJ, François A, Cauliez A, Zhukouskaya VV, Linglart A, Chaussain C, Bardet C. JBMR Plus. 2026 Jan 28;10(4):ziag011. doi: 10.1093/jbmrpl/ziag011. eCollection 2026 Apr.
XLH : que se passe-t-il quand le burosumab s’arrête à la fin de la croissance ?
C’est tout l’intérêt de l’étude MyXLH, qui s’est attachée à documenter, au plus près du vécu des patients, ce qui change ou non dans les mois qui suivent l’arrêt ou la poursuite du traitement. Pour cela, les chercheurs ont combiné données biologiques, symptômes rapportés via application mobile, activité physique enregistrée par objet connecté, questionnaires de qualité de vie et entretiens téléphoniques approfondis.
L’étude a inclus 25 adolescents suivis dans plusieurs centres européens. Tous avaient reçu du burosumab avant la fin de leur croissance, puis ont été observés pendant 26 semaines après cette étape. Chez ceux qui ont poursuivi le traitement, les auteurs décrivent une phosphatémie globalement stable, des scores de douleur, raideur et fatigue restant faibles, ainsi qu’un niveau d’activité physique globalement maintenu. Dans les entretiens, ces adolescents racontent surtout une vie quotidienne relativement préservée : école, loisirs, activités physiques, relations sociales, avec un mieux-être émotionnel souvent lié à l’amélioration des symptômes.
Le tableau est plus contrasté chez les adolescents qui ont arrêté le burosumab à la fin de la croissance. Dans ce groupe, la phosphatémie redescendait sous les normes, tandis que la douleur, la raideur et la fatigue avaient tendance à augmenter, même si l’intensité restait variable d’un patient à l’autre. Les entretiens donnent ici toute leur profondeur aux chiffres : certains adolescents décrivent des douleurs nouvelles ou plus marquées, une gêne accrue pour marcher, courir ou travailler, et parfois un retentissement sur les activités scolaires, sociales ou sur l’humeur. Tous ne vivaient pas cette transition de la même façon, mais plusieurs rapportaient nettement la perte d’un bénéfice qu’ils associaient au traitement. De quoi renforcer l’idée que, chez une partie de ces patients, la poursuite du traitement au-delà de la fin de croissance mérite d’être envisagée pour maintenir le contrôle des symptômes et préserver la qualité de vie.
Pour en savoir + : Adolescents' experience of living with X-linked hypophosphataemia (XLH): a mixed-methods analysis of those who continued and discontinued burosumab treatment after end of skeletal growth.
Saraff V, Arango-Sancho P, Bacchetta J, Boot AM, Burren CP, Chinoy A, Dharmaraj P, Gómez Llorente MA, González Rodríguez JD, Gueorguieva I, Hayes W, Schnabel D, Duro HR, Davies EH, Komarzynski S, Rylands AJ, Sandilands K, Ishii H, Williams A, Selveindran S, Barlassina A, Bowden A, Linglart A. Orphanet J Rare Dis. 2026 Feb 3;21(1):107. doi: 10.1186/s13023-026-04244-2.
Hypophosphatasie : quand la biologie parle avant les symptômes
Certaines personnes porteuses d’un variant du gène ALPL présentent les marqueurs biologiques typiques de l’hypophosphatasie, sans aucun signe clinique. Cette étude identifie 43 sujets dans cette situation et propose de mieux reconnaître ce profil particulier, pour éviter à la fois le sous-diagnostic… mais aussi le surdiagnostic.
L’hypophosphatasie (HPP) est une maladie génétique rare liée à des anomalies du gène ALPL, responsable d’une baisse de l’activité de la phosphatase alcaline. Cette déficience enzymatique peut entraîner une accumulation de certains substrats, notamment le PLP (la forme circulante de la vitamine B6) et/ou le PEA urinaire, deux marqueurs biologiques classiquement associés à la maladie. Mais l’expression clinique est très variable.
À partir de la base internationale des variants ALPL, les chercheurs ont identifié 43 individus asymptomatiques porteurs d’au moins un variant ALPL et présentant tous une phosphatase alcaline basse pour l’âge et le sexe. Parmi les sujets pour lesquels les substrats de l’enzyme étaient documentés, plusieurs avaient aussi un PLP et/ou un PEA élevés, renforçant le profil biochimique compatible avec l’hypophosphatasie. Aucun ne présentait toutefois de signe clinique ou radiologique rapporté au moment de l’analyse.
La majorité de ces sujets étaient hétérozygotes (âge médian 29 ans), mais l’étude montre surtout une forte variabilité génotype-phénotype : certains génotypes observés dans cette cohorte asymptomatique ont déjà été décrits dans des formes odontologiques, infantiles, juvéniles ou adultes d’hypophosphatasie. La matrice de la page 7 illustre bien cette diversité d’expression pour des génotypes parfois identiques. Les auteurs proposent donc de considérer ces situations comme un phénotype biochimique d’hypophosphatasie chez des porteurs asymptomatiques de variants ALPL, plutôt que comme une hypophosphatasie clinique avérée. Ils soulignent aussi que l’évolution de ces profils reste inconnue : certains pourraient rester asymptomatiques, d’autres éventuellement développer des manifestations plus tardives. D’où l’intérêt d’un suivi raisonné, sans conclure trop vite à une maladie exprimée ni envisager un traitement systématique.
Pour en savoir + : Biochemical phenotype of hypophosphatasia in asymptomatic individuals carrying ALPL variants.
Montero-Lopez R, Farman MR, Högler F, Rehder C, Malli T, Webersinke G, Rockman-Greenberg C, Dahir K, Martos-Moreno GÁ, Linglart A, Ozono K, Seefried L, Del Angel G, Nading EB, Huggins E, Rush ET, Tauer JT, Kishnani PS, Högler W.J Bone Miner Res. 2026 Mar 2;41(3):259-269. doi: 10.1093/jbmr/zjaf124.
Hypophosphatasie : des résultats de phase III contrastés pour l’efzimfotase alfa
Dans un communiqué publié le 31 mars 2026, AstraZeneca/Alexion font état de résultats globalement encourageants pour l’efzimfotase alfa dans l’hypophosphatasie, à l’issue d’un programme mondial de phase III mené chez 196 patients dans 22 pays. Le signal apparaît particulièrement favorable en pédiatrie, tandis que les données apparaissent plus contrastées chez les adolescents et les adultes.
Chez les enfants n’ayant jamais reçu d’asfotase alfa, l’essai MULBERRY a atteint son critère d’évaluation principal. L’efzimfotase alfa a permis, à 25 semaines, une amélioration statistiquement significative et cliniquement pertinente de la santé osseuse, évaluée par le Radiographic Global Impression of Change (RGI-C), ainsi qu’un bénéfice sur la sévérité du rachitisme. Les autres critères secondaires, notamment ceux liés à la fonction physique et à la motricité, vont dans le sens d’un bénéfice clinique global chez l’enfant.
MULBERRY était un essai de phase III, randomisé, en double aveugle, contre placebo, mené chez des enfants de 2 à moins de 12 ans, avec 29 patients recrutés dans 14 pays. Pour être éligibles, les patients devaient présenter une HPP avec rachitisme visible à la radiographie, un faible taux d’ALP (phosphatase alcaline), ainsi qu’un variant du gène ALPL ou une élévation du PLP (pyridoxal-5’-phosphate). Les participants recevaient de l’efzimfotase alfa ou un placebo toutes les deux semaines pendant 24 semaines.
Chez les enfants déjà traités par l’asfotase alfa, l’essai CHESTNUT rapporte une bonne tolérance et le maintien du bénéfice thérapeutique après switch vers l’efzimfotase alfa. En revanche, chez les adolescents et adultes naïfs de l’asfotase alfa, l’essai HICKORY n’a pas atteint son critère principal sur le test de marche de 6 minutes, malgré une amélioration numérique. Le communiqué mentionne néanmoins des signaux favorables sur la fatigue dans l’ensemble de la population étudiée et, dans des sous-groupes prédéfinis de patients atteints d’une HPP à début pédiatrique, sur la mobilité, la fonction physique et la douleur.
À ce stade, ces données proviennent d’un communiqué industriel et doivent encore être présentées en congrès scientifique et soumises aux autorités réglementaires.
Hypoparathyroïdie chronique : sortir du prisme de la seule hypocalcémie
Récemment publiée dans Trends in Endocrinology & Metabolism (Cell Press), une revue co-signée notamment par Jean-Philippe Bertocchio (CCMR des maladies rares du métabolisme du calcium et du phosphate, hôpital de la Pitié-Salpêtrière, AP-HP) invite à revoir la manière de comprendre l’hypoparathyroïdie chronique. Cette pathologie ne se résume pas à une simple hypocalcémie, mais correspond à un déficit fonctionnel beaucoup plus large.
Au-delà du calcium, une hormone qui manque à plusieurs étages
Sur le papier, le traitement conventionnel paraît simple : calcium et vitamine D active pour corriger la calcémie. En pratique, la revue montre pourquoi cette stratégie reste incomplète. Elle corrige certains déséquilibres biologiques, sans pour autant reproduire le rôle naturel de la PTH au niveau du rein, de l’os, et indirectement de l’intestin. Conséquence : même bien traités, de nombreux patients continuent à présenter des symptômes persistants, de variations biologiques et de complications à long terme, malgré un traitement bien conduit.
La PTH agit normalement comme un chef d’orchestre de l’homéostasie phosphocalcique. Elle augmente la réabsorption rénale du calcium et du magnésium, stimule la synthèse rénale de vitamine D active, limite la réabsorption du phosphate et régule en continu le remodelage osseux. Lorsqu’elle fait défaut, la baisse du calcium sanguin n’est donc qu’une partie du tableau. S’y ajoutent fréquemment une hyperphosphatémie, un ralentissement du remodelage osseux, une hausse de l’excrétion urinaire du calcium et un ensemble de conséquences fonctionnelles qui dépassent largement le simple métabolisme minéral.
Une maladie qui se ressent dans tout le corps
La revue souligne l’importance du fardeau symptomatique. Le déficit chronique en PTH peut entraîner des manifestations neuromusculaires comme les crampes, les spasmes, les paresthésies, la faiblesse musculaire ou la tétanie. Mais il peut aussi s’accompagner d’atteintes cardiovasculaires, neuropsychiatriques, ophtalmologiques, dentaires, cutanées, respiratoires ou immunitaires. Certaines complications cardiaques (arythmies, insuffisance cardiaque, événements ischémiques) seraient liées non seulement à l’hypocalcémie, mais aussi aux fluctuations de la calcémie et à l’excès de phosphate. Cet impact se ressent fortement au quotidien. Malgré le traitement conventionnel, les patients rapportent souvent une qualité de vie altérée, avec des répercussions sur le sommeil, l’activité physique, la vie familiale et les tâches courantes. Les études citées mentionnent également des troubles cognitifs objectivables, notamment au niveau de l’attention, de la vitesse de traitement de l’information et des fonctions exécutives.
Le rein, victime silencieuse
Le retentissement rénal n’est pas un effet secondaire anecdotique : en l’absence de PTH, le rein réabsorbe moins bien le calcium. Si l’on ajoute à cela de fortes doses de calcium oral et de vitamine D active, on peut aboutir à un état de surcharge calcique urinaire, autrement dit à une hypercalciurie, susceptible d’endommager progressivement le rein. En effet, il a été rapporté qu’environ 36 à 40 % des patients présentent une hypercalciurie ; 10 à 31 % développent une néphrocalcinose ou une néphrolithiase ; et le risque de maladie rénale chronique est rapporté comme 2 à 3 fois plus élevé que chez les témoins, avec des estimations encore plus marquées dans certaines cohortes. Le schéma ci-dessous résume bien la cascade proposée : déficit en PTH, traitement conventionnel à fortes doses, excès de calcium urinaire, dépôts calciques, altération rénale puis progression possible vers la maladie rénale chronique.
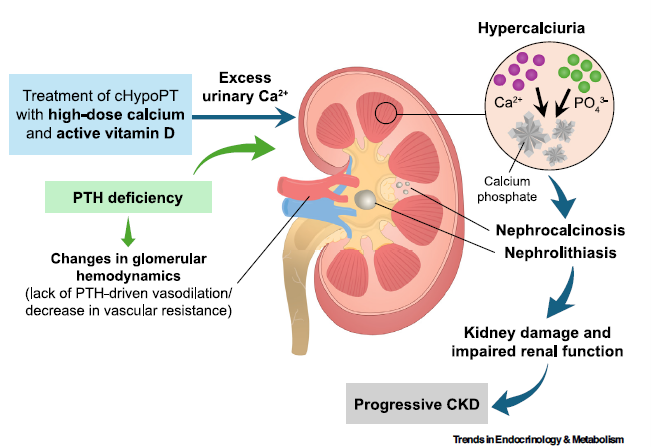
Figure tirée de Brandi M and al. Trends in Endocrinology & Metabolism DOI: (10.1016/j.tem.2026.03.009)
De surcroit, les auteurs suggèrent que l’absence de PTH pourrait avoir un effet délétère propre sur le rein, indépendamment des dépôts calciques, en modifiant l’hémodynamique glomérulaire. Ainsi, le rein souffrirait à la fois du déséquilibre minéral et de la perte d’un signal hormonal protecteur.
Le paradoxe osseux : une densité élevée ne protège pas forcément
Pendant longtemps, l’hypoparathyroïdie chronique a été perçue comme une situation plutôt « protectrice » pour l’os, en raison d’une densité minérale osseuse souvent plus élevée que la moyenne. Mais cette densité n’est pas synonyme de solidité garantie. Ce que l’on gagne parfois en masse osseuse pourrait se payer en qualité osseuse et en moindre capacité d’adaptation mécanique.
Les auteurs décrivent une forte hétérogénéité des profils osseux. Chez certains patients, en particulier les femmes ménopausées, les hommes de 50 ans et plus et les patients ayant une forme génétique, l’ostéopénie, l’ostéoporose et les fractures de fragilité sont loin d’être rares. La figure ci-dessous illustre parfaitement ce changement de paradigme : d’un côté, la vision d’un os dense à remodelage ralenti ; de l’autre, des sous-groupes chez qui apparaissent ostéoporose, dégradation microarchitecturale et fractures vertébrales.
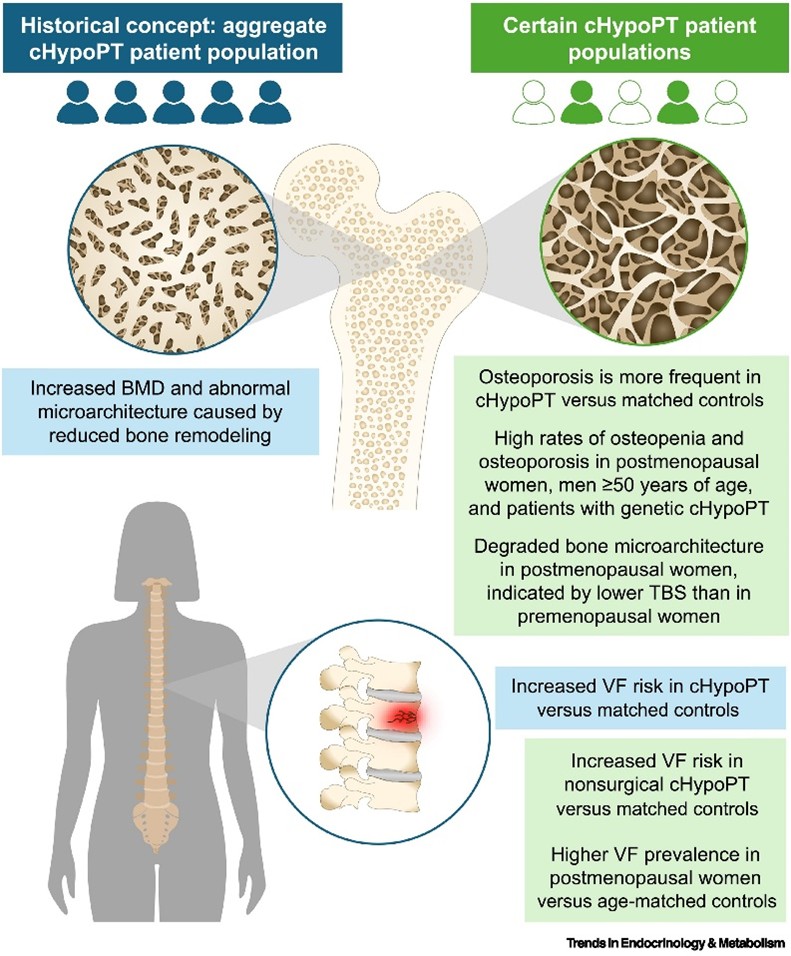
Figure tirée de Brandi M and al. Trends in Endocrinology & Metabolism DOI: (10.1016/j.tem.2026.03.009)
Fractures vertébrales : le vrai signal d’alerte
L’hypoparathyroïdie chronique ne protège pas contre la fragilité osseuse. Plusieurs études citées par la revue convergent vers une augmentation du risque de fractures vertébrales, en particulier chez certains sous-groupes. Une grande cohorte nationale suédoise a ainsi retrouvé un sur-risque de fracture vertébrale chez les patients atteints d’hypoparathyroïdie chronique, même si le risque fracturaire n’évoluait pas de façon uniforme selon les sites osseux.
Les auteurs insistent sur la nécessité d’évaluer non seulement la densité, mais aussi la microarchitecture osseuse, par exemple via le trabecular bone score (TBS), ainsi que le contexte clinique global. Chez les femmes ménopausées, plusieurs études rapportent un TBS plus bas que chez les femmes préménopausées, signe d’une microarchitecture plus dégradée malgré une densité parfois conservée, voire augmentée.
Un coût humain, social et économique encore sous-estimé
L’article ne s’arrête pas à la physiopathologie. Il rappelle aussi que cette maladie pèse lourd sur le plan social et économique. Les patients ont davantage recours aux soins, avec notamment environ deux fois plus d’hospitalisations, davantage de passages aux urgences et de consultations. Sur le plan professionnel, les enquêtes citées montrent un impact important sur la capacité de travail, la productivité et parfois le maintien dans l’emploi.
À cela s’ajoute la charge invisible : fatigue, symptômes chroniques, troubles cognitifs, anxiété liée aux complications, dépendance à un traitement lourd et surveillance répétée. La revue rappelle ainsi que l’hypoparathyroïdie chronique n’est pas seulement une maladie « biologique » ; c’est aussi une maladie du fonctionnement quotidien.
Vers un changement de stratégie thérapeutique ?
Tant que l’on traite uniquement la calcémie, on laisse de côté une part essentielle de la maladie. Les auteurs soulignent donc l’intérêt croissant des thérapies de remplacement par la PTH chez les patients insuffisamment contrôlés par le traitement conventionnel. De plus, la prise en charge devrait être plus personnalisée, avec une attention renforcée aux patients à risque rénal et aux sous-groupes osseux fragiles, notamment les femmes ménopausées, les hommes plus âgés et les formes génétiques
Pour en savoir + : Understanding hypoparathyroidism as a disease of broad functional deficiency.
Maria Luisa Brandi, Jean-Philippe Bertocchio, Istvan Takacs, Ellen K. Colman, Ying Kong, Lars Rejnmark, Aliya A. Khan. Trends in Endocrinology & Metabolism, Month 2026, Vol. xx, No. xx https://doi.org/10.1016/j.tem.2026.03.00
L’hypoparathyroïdie : quand l’os retrouve son rythme ?
Pendant longtemps, dans l’hypoparathyroïdie chronique, l’urgence a été de compenser ce que l’organisme ne produisait plus : la parathormone. En pratique, cela revenait surtout à administrer du calcium et de la vitamine D active pour maintenir une calcémie acceptable. Une stratégie efficace pour limiter les symptômes, mais qui ne remplace pas vraiment les multiples fonctions de la PTH. Car cette hormone ne se contente pas de réguler le calcium dans le sang : elle agit aussi sur le rein, l’intestin… et l’os. Or, sans elle, le squelette entre dans une forme de ralentissement. Son remodelage diminue, sa densité augmente souvent, mais cette densité plus élevée n’est pas nécessairement synonyme d’un os plus « normal ».
C’est précisément cette dimension longtemps négligée qu’a explorée une équipe internationale chez des adultes atteints d’hypoparathyroïdie chronique traités par palopegteriparatide, une PTH (1-34) conçue pour être libérée progressivement pendant 24 heures.
Les chercheurs ont suivi 59 patients pendant trois ans dans le cadre de l’essai clinique PaTH Forward. Sous traitement, les marqueurs biologiques du remodelage osseux augmentent rapidement, puis se stabilisent, tandis que la densité minérale osseuse (DMO) diminue modérément vers des valeurs plus physiologiques.
De surcroit, à 162 semaines, la calcémie et la calciurie restent dans les normes, et 91 % des patients n’ont plus besoin du traitement conventionnel par vitamine D active et fortes doses de calcium. Autrement dit, ce traitement ne se contente pas de corriger la biologie, il pourrait aussi redonner au squelette une part de son rôle naturel dans l’équilibre du calcium.
Pour en savoir + : Palopegteriparatide for Adults with Chronic Hypoparathyroidism: Skeletal Dynamics Through 3 yr of the Phase 2 paTH Forward Trial.
Rubin MR, Clarke BL, Hofbauer LC, Khan A, Schwarz P, Vokes T, Ahmed I, Palermo A, Cetani F, Pagotto U, Zhao C, Ominsky MS, Lai B, Ukena J, Shu AD, Rejnmark L.J Bone Miner Res. 2026 Feb 4:zjag013. doi: 10.1093/jbmr/zjag013.
Quand la pompe prend le relais dans l’hypoparathyroïdie
Selon une récente étude menée dans trois centres de référence des maladies rares du métabolisme du calcium et du phosphate (CaP), l’administration continue de tériparatide par pompe sous-cutanée dans le traitement de l’hypoparathyroïdie chronique pourrait offrir une nouvelle option aux patients qui restent insuffisamment contrôlés malgré les traitements habituels. Chez ces malades, la stratégie semble améliorer certains paramètres biologiques et alléger le traitement oral, sans toutefois répondre encore à toutes les questions, notamment sur la qualité de vie et l’os à long terme.
Pour en savoir + : Continuous subcutaneous administration of teriparatide as a therapeutic option in chronic hypoparathyroidism: retrospective study in three tertiary care centers of the epi-hypo study.
Attia A, Brière M, Kamenicky P, Lecoq AL, Maione L, Houillier P, Buffet C, Bertocchio JP, Ghander C.Eur J Endocrinol. 2026 Jan 6;194(1):K1-K7. doi: 10.1093/ejendo/lvaf257.
Cystinose : la thérapie génique entre en clinique
Chez six adultes atteints de cystinose, une thérapie génique à partir de cellules souches hématopoïétiques autologues montre un profil de sécurité jugé acceptable et un signal biologique encourageant sur la surcharge en cystine. Des résultats encore préliminaires, mais qui ouvrent une piste de traitement !
Dans la cystinose, les mutations du gène CTNS empêchent le bon fonctionnement de la cystinosine, transporteur lysosomal de la cystine. Cette dernière s’accumule dans les cellules de l’ensemble de l’organisme. Sur le plan rénale, la maladie débute précocement par un syndrome de Fanconi, puis évolue vers l’insuffisance rénale chronique et souvent vers l’insuffisance rénale terminale. La cystéamine ralentit cette progression, sans l’empêcher totalement. D’où l’intérêt d’approches capables de restaurer durablement une fonction cellulaire normale.
L’étude publiée dans le New England Journal of Medicine a évalué une stratégie de thérapie génique ex vivo chez six patients âgés de 20 à 46 ans. Le principe : prélever des cellules souches CD34+ autologues, les modifier à l’aide d’un vecteur lentiviral portant un ADN complémentaire de CTNS, puis les réinjecter après conditionnement par busulfan. Tous les patients ont été suivis entre 29 et 63 mois.
Sur le plan de la tolérance, 216 événements indésirables ont été rapportés, majoritairement légers ou modérés, et globalement compatibles avec le conditionnement, la procédure de greffe ou la maladie elle-même.
Sur le plan biologique, les taux de cystine dans les globules blancs ont diminué chez la plupart des patients, ce qui va dans le sens d’un effet du traitement. Chez les patients déjà greffés du rein, la fonction rénale est restée globalement stable. Mais chez les deux patients qui avaient déjà une insuffisance rénale chronique modérée au départ, l’évolution n’a pas été stoppée : l’un a vu sa fonction rénale se dégrader jusqu’au stade terminal, et l’autre a présenté une baisse importante de son débit de filtration glomérulaire, c’est-à-dire de la capacité de ses reins à filtrer le sang. Autrement dit, l’étude apporte une preuve de concept biologique, mais pas encore la démonstration d’un bénéfice rénal clinique chez des adultes déjà avancés dans l’histoire naturelle de la maladie.
Pour en savoir + : Hematopoietic Stem-Cell Gene Therapy for Cystinosis.
Barshop BA, Ball ED, Benador N, Trauner D, Phillips S, Dohil R, Afshari NA, Roy S, Campo Fernandes B, Kohn D, Shayan K, Everett JK, Bushman FD, Midgley J, Liang H, Sawyers A, Gangoiti JA, Panchal M, Ahmed I, Cherqui S.N Engl J Med. 2026 Feb 19;394(8):753-762. doi: 10.1056/NEJMoa2506431.


