Actu CAP - Janvier 2026

Sommaire
🔎 Recherche clinique : les “basket trials” à l’épreuve des maladies rares
🔎 Hyperpara modéré, hypopara chronique, grossesse : les parathyroïdes au crible des experts européens
🔎 Étude CALIBRATE dans l’hypocalcémie autosomique dominante
🔎 iPPSD3 : des variants d’épissage de GNAS dérèglent l’empreinte génomique
🔎 Rachitismes hypophosphatémiques et ostéomalacie : des nouveautés
Recherche clinique : les “basket trials” à l’épreuve des maladies rares
Alors que plus de 95 % des maladies rares restent dépourvues de traitements spécifiques, la recherche clinique est confrontée à un paradoxe : un besoin thérapeutique immense face à une faisabilité méthodologique limitée. Dans une revue systématique d’envergure, Wael Khazen et al. examinent l’émergence des “basket trials”, une approche innovante qui pourrait transformer le développement de thérapies ciblées dans les maladies rares.
Une nouvelle façon de tester les médicaments dans les maladies rares
Alors que la majorité des 7 000 maladies rares identifiées ne disposent d’aucun traitement spécifique, la mise au point de nouveaux médicaments se heurte à des obstacles bien connus : effectifs patients très faibles, grande hétérogénéité clinique, absence de biomarqueurs validés et critères d’évaluation peu standardisés. Dans ce contexte, les basket trials proposent une réponse originale : regrouper, au sein d’un même protocole, des patients atteints de maladies différentes mais partageant une anomalie biologique ou génétique commune, pour évaluer un traitement ciblé. Ce changement de paradigme, centré sur le mécanisme plutôt que sur la maladie, s’inscrit dans la logique plus large de la médecine de précision.
36 essais analysés, 9 hors oncologie
L’analyse de 36 essais identifiés par les auteurs montre que cette méthodologie est encore largement dominée par l’oncologie (75 % des cas), où l’organisation par mutations (comme BRAF ou NTRK) est bien établie. Dans les maladies rares non cancéreuses, seuls neuf essais ont été recensés. Ils couvrent 25 pathologies distinctes sans chevauchement, soulignant à la fois le potentiel du modèle et sa faible maturité hors du champ oncologique. Les designs restent majoritairement non randomisés et ouverts, avec des durées d’études souvent longues (plus de six ans en moyenne) et une charge logistique importante, parfois répartie sur des centaines de centres.
Mais ces projets restent confrontés à des limites structurelles. Dans les maladies rares non tumorales, les critères de jugement sont hétérogènes et rarement comparables d’un bras à l’autre. Les biomarqueurs manquent, les populations sont encore plus dispersées, et les contraintes éthiques, notamment en pédiatrie, restreignent les possibilités de randomisation ou de bras contrôle. Ainsi, malgré des avancées notables, le basket trial reste aujourd’hui surtout un outil exploratoire, sous-utilisé au-delà de l’oncologie.
Vers une évolution du modèle : recommandations clés
Les auteurs appellent donc à un changement d’échelle et de méthode. Pour que ces essais deviennent une voie structurante du développement thérapeutique dans les maladies rares, ils recommandent une meilleure harmonisation des critères d’évaluation, un recours plus large aux méthodes statistiques adaptatives (designs bayésiens, contrôles synthétiques) et une implication renforcée des patients, notamment via les réseaux européens (ERNs, ERICA, EU-X-CT) et les plateformes de données partagées.
En somme, les basket trials ne sont pas une solution miracle. Mais s’ils sont bien conçus, avec rigueur scientifique et intelligence collective, ils peuvent permettre d’accélérer significativement l’accès aux traitements pour des patients longtemps laissés sans solution. C’est un chantier autant méthodologique que politique, qui appelle une mobilisation transversale : chercheurs, cliniciens, autorités de régulation, industriels, mais aussi patients et familles.
Pour en savoir + : Basket trials in rare diseases: a systematic review of current practices, methodological challenges, and future directions.
Khazen W, Corriol-Rohou S, Evangelista T, Valent A, Abbas S, Nissan X, Mejat A.Orphanet J Rare Dis. 2025 Nov 12;20(1):578.
Hyperpara modéré, hypopara chronique, grossesse : les parathyroïdes au crible des experts européens
À l’occasion du workshop européen PARAT organisé en mai 2024 par la Société Européenne d’Endocrinologie (ESE), un groupe multidisciplinaire d’experts a dressé un état des lieux clinique actualisé de trois grands volets de la pathologie parathyroïdienne : l’hyperparathyroïdie primaire (PHPT) modérée ou asymptomatique, l’hypoparathyroïdie chronique (HypoPTH) et les troubles du métabolisme calcique liés à la grossesse. Ce rapport structuré autour de vignettes cliniques et de recommandations pratiques propose une synthèse à fort impact pour la pratique courante.
Hyperparathyroïdie légère : entre attente et scalpel
La PHPT est de plus en plus souvent diagnostiquée chez des patients asymptomatiques ou pauci-symptomatiques, notamment via le dosage de la calcémie. Le bénéfice d’une chirurgie parathyroïdienne dans les formes modérées reste débattu : si la densité osseuse (DMO) rachidienne s’améliore post-chirurgie, le lien avec une réduction du risque de fractures reste incertain.
Sur le plan rénal, les auteurs rappellent que seule l’hypercalcémie franche est délétère. L’amélioration de la qualité de vie post-parathyroïdectomie est quant à elle modeste. En pratique, une surveillance est privilégiée en l’absence de critères classiques opératoires, en particulier chez les personnes âgées.
Hypoparathyroïdie chronique : la substitution par PTH devient réalité
L’arrivée du palopegteriparatide (agoniste du récepteur de la PTH à demi-vie prolongée) marque un tournant dans la prise en charge des patients avec HypoPTH difficile à équilibrer. Le rapport détaille les critères de sélection, la transition thérapeutique (avec arrêt progressif du calcium et de l’alfacalcidol), et les paramètres biologiques à surveiller (calcémie, phosphatémie, calciurie, fonction rénale). Les auteurs insistent sur le besoin de structurer un suivi expert et d’évaluer finement l’impact à long terme, notamment sur l’os et la fonction rénale (voir article ci-dessous).
Troubles parathyroïdiens en période périnatale : repères pratiques
Le rapport consacre un chapitre spécifique aux troubles du métabolisme phosphocalcique pendant la grossesse et l’allaitement. L’hyperparathyroïdie pendant la gestation, bien que rare, peut engager le pronostic materno-fœtal. La chirurgie au deuxième trimestre peut s’imposer, notamment en cas d’hypercalcémie menaçante.
À l’opposé, l’HypoPTH expose à des fluctuations calciques à risque pour le fœtus (hypocalcémie néonatale, retard de croissance) : un algorithme pragmatique de surveillance et d’adaptation thérapeutique est proposé. Quelques cas sous traitement par PTH pendant la grossesse sont également évoqués, sans signal de sécurité défavorable.
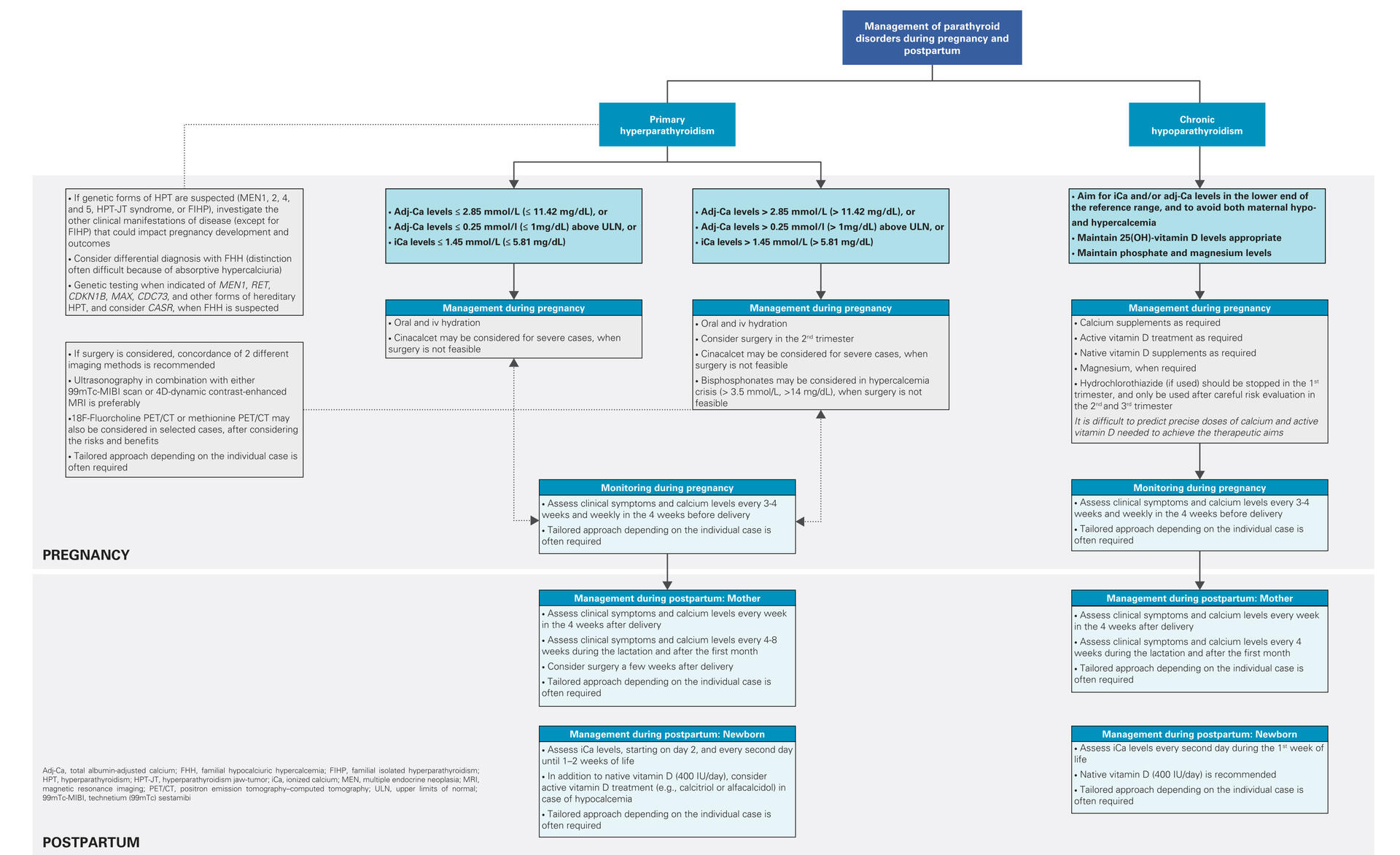
Figure : Prise en charge des troubles parathyroïdiens pendant la grossesse et après l'accouchement. D’après Luís Miguel Cardoso et al. European Journal of Endocrinology, 2025.
Focus génétique et syndromes rares
Le rapport souligne l’importance du dépistage moléculaire dans les formes familiales ou syndromiques , en particulier dans les néoplasies endocriniens multiples (NEM), ou les formes juvéniles avec hypercalcémie persistante. Le choix du type de chirurgie, notamment pour préserver la fonction parathyroïdienne, doit être guidé par l'identification génétique et concerté en réunion pluridisciplinaire.
L’atelier PARAT 2024 remet au centre du jeu une médecine personnalisée dans les troubles des parathyroïdes, à la croisée de l’endocrinologie, de la chirurgie, de la néphrologie et de l’obstétrique. Les évolutions récentes (traitement substitutif par PTH, indications chirurgicales plus fines, prise en charge périnatale) obligent à une mise à jour des pratiques et à une structuration des parcours de soins autour des centres référents.
Pour en savoir + : Advances in the clinical management of parathyroid disorders: report from the 2024 workshop by the ESE educational program on parathyroid disorders.
Cardoso LM, Rolighed L, Amrein K, Pilz S, Underbjerg L, Pretorius M, Cetani F, Zahn A, Almquist M, Makay O, Marcocci C, Rejnmark L, Siggelkow H, Tsourdi E, Kamenický P, Bollerslev J.Eur J Endocrinol. 2025 Nov 26;193(6):R65-R88
L’hypoparathyroïdie chronique : l’ESE révise sa stratégie de prise en charge !
La Société Européenne d’Endocrinologie (ESE) a publié en 2025 une version révisée de son guide de pratique clinique sur l’hypoparathyroïdie chronique de l’adulte. Cette révision était motivée par de nouvelles données publiées depuis 2015 sur le fardeau de la maladie, la qualité de vie des patients et les comorbidités associées, ainsi que par la reconnaissance des limites du traitement conventionnel au long cours.
En effet, les patients porteurs d’une hypoparathyroïdie présentent une qualité de vie dégradée (douleurs musculaires, fatigue, altération du bien-être psychique) et un risque accru de troubles neurologiques (dépression, anxiété), rénaux (insuffisance rénale, lithiases) et cardiovasculaires (maladie coronarienne, calcifications). De plus, les traitements usuels (calcium et analogues de vitamine D) exigent une prise médicamenteuse élevée et exposent au risque d’hypercalcémie et d’hypercalciurie, susceptibles d’engendrer des complications rénales. Ce constat a conduit le comité d’experts, incluant des endocrinologues de plusieurs pays européens, à actualiser les recommandations sur la base des données les plus récentes, malgré la rareté des essais cliniques disponibles.
Changements clés depuis 2015
- Définition revisitée : une hypoparathyroïdie post-chirurgicale est désormais qualifiée de chronique au-delà de 12 mois (contre 6 mois précédemment), en raison de possibles récupérations tardives de la fonction parathyroïdienne.
- Diagnostic génétique encouragé : en l’absence de cause évidente, une analyse génétique est recommandée (ex. mutations de CASR, PTH, GCM2), y compris en cas d’absence d’antécédent familial.
- Traitement conventionnel mieux encadré : l’objectif thérapeutique est de maintenir une calciémie basse-normale sans hypercalciurie, avec un schéma reposant sur calcitriol ou alfacalcidol + calcium, en privilégiant une titration lente.
- Nouveaux critères pour la PTH recombinante : le guide reconnaît le rôle élargi de la substitution hormonale chez les patients insuffisamment équilibrés ou symptomatiques malgré le traitement classique, notamment en cas :
- de qualité de vie altérée,
- d’insuffisance rénale modérée,
- d’hyperphosphatémie ou d’hypercalciurie persistantes.
- Approbation de la PTH à longue durée d’action : le palopegteriparatide, désormais autorisée en Europe (EMA, 2023), constitue une option de premier plan avec une seule injection quotidienne, une réduction de la charge médicamenteuse et une amélioration des paramètres biochimiques (phosphate, calciurie, etc.).
- Rejet de la greffe parathyroïdienne : les données actuelles ne permettent pas de recommander l’allotransplantation comme option thérapeutique.
- Suivi personnalisé et contextuel : le guide précise les modalités de surveillance biologique, la gestion spécifique en cas de grossesse, allaitement, insuffisance rénale ou formes génétiques.
Les auteurs soulignent que les preuves restent « très faibles » pour de nombreux critères (petits effectifs, études observationnelles). Ils pointent notamment le manque d’essais contrôlés randomisés portant sur les issues cliniques (mortalité, qualité de vie, complications rénales et osseuses).
Les effets à long terme des nouvelles thérapies PTH sur la mortalité, le risque cardiovasculaire, les fractures et la fracture de qualité de vie nécessitent des études supplémentaires. De même, les données sont insuffisantes sur l’optimisation de la calcémie cible et sur le rôle exact de la calcitonine-like peptide dans cette pathologie rare. La place de la transplantation parathyroïdienne reste incertaine, tout comme le bénéfice des nouvelles molécules en développement (PTH oral, calcilytiques, agents sélectifs). La restauration tardive de la fonction parathyroïdienne au-delà d’un an est rare mais possible, suggérant qu’un suivi prolongé reste nécessaire. Ces zones d’ombre justifient une recherche continue pour améliorer la prise en charge de l’HypoPTH adulte.
Pour en savoir + : Revised European Society of Endocrinology Clinical Practice Guideline: Treatment of Chronic Hypoparathyroidism in Adults.
Bollerslev J, Buch O, Cardoso LM, Gittoes N, Houillier P, van Hulsteijn L, Makay O, Marcocci C, Pallais JC, Pilz S, Rejnmark L, Yavropoulou M, Dekkers OM.
Étude CALIBRATE dans l’hypocalcémie autosomique dominante
Cette forme héréditaire d’hypoparathyroïdie due à une mutation activatrice du CaSR a bénéficié des avancées avec l’encaleret. Il a en effet réussi un essai de phase III (étude CALIBRATE) chez ces patients atteints d’hypocalcémie autosomique dominante de type 1 (ADH1). Les résultats publiés fin 2025 montrent que 76 % des patients traités ont atteint simultanément une calcémie et une calciurie normales à 24 semaines, contre seulement 4 % sous traitement conventionnel (calcium/vitamine D).
De plus, 91 % des patients sous encaleret ont retrouvé un taux de PTH endogène dans la norme. Aucune interruption pour effet indésirable n’a été rapportée. Ces données « remarquables » selon les endocrinologues impliqués confirment le potentiel de ce traitement oral ciblé à devenir un nouveau standard de soin pour l’ADH1. Une demande d’AMM est prévue au 1er semestre 2026, et des études chez l’enfant et dans l’hypoparathyroïdie chronique sont programmées.
iPPSD3 : des variants d’épissage de GNAS dérèglent l’empreinte génomique
La pseudohypoparathyroïdie de type 1B (PHP1B), ou iPPSD3 est liée à une diminution d’expression de Gsα secondaire à des anomalies d’empreinte (méthylation) au locus GNAS. Si certaines formes autosomiques dominantes sont expliquées par des délétions (ex. STX16), la cause génétique de la forme la plus fréquente avec perte de méthylation étendue (Cat1) reste souvent inconnue. Les auteurs identifient pour la première fois, dans une famille avec deux sœurs atteintes, un variant nucléotidique au site donneur d’épissage de l’exon H de GNAS (chr20:58,841,877 T>C), absent des grandes bases de données, et compatible avec un effet “parent-of-origin”. Des expériences in vitro (minigène dans des cellules souches embryonnaires humaines et éditions CRISPR) montrent que ces variants entraînent un épissage aberrant et une atténuation de la transcription issue de l’exon H, mécanisme proposé pour empêcher l’établissement des marques de méthylation maternelles durant l’ovogenèse.
Cette découverte éclaire d’un jour nouveau la pathogenèse des formes sporadiques de PHP1B, dont les causes génétiques restaient inconnues dans de nombreux cas.
Côté thérapeutique, bien qu’aucun traitement spécifique de la résistance au PTH ne soit encore disponible, des pistes émergent : par exemple, un essai clinique de phase II est en cours pour évaluer l’effet de la théophylline (un inhibiteur de phosphodiestérase) dans la pseudohypoparathyroïdie, afin d’augmenter les niveaux d’AMPc intracellulaire et potentiellement contourner la résistance hormonale.
Pour en savoir + : Defective GNAS imprinting due to splice site variants in pseudohypoparathyroidism type 1B.
Iwasaki Y, Reyes M, Molin A, Muurinen M, Kottler ML, Bastepe M, Jüppner H.JCI Insight. 2025 Sep 2;10(19):e194754.
Rachitismes hypophosphatémiques et ostéomalacie : des nouveautés
En septembre 2025, la FDA a approuvé une extension d’indication de burosumab au traitement de l’ostéomalacie oncogénique (TIO, tumor-induced osteomalacia) chez l’adulte et l’enfant à partir de 2 ans, dans les cas où la tumeur sécrétant le FGF23 ne peut être localisée ou réséquée. Cette décision fait suite aux données des études de phase II montrant que burosumab corrige la phosphatémie et améliore la minéralisation osseuse même dans ces formes paranéoplasiques rares. Par ailleurs, au Japon, une nouvelle présentation de burosumab en seringue pré-remplie a été lancée fin 2025 pour faciliter l’auto-administration du traitement. Cette formulation prête à l’emploi, approuvée en juin 2025, réduit les manipulations et vise à alléger la charge thérapeutique des patients et leurs familles.
XLH Matters 2024
Le congrès XLH Matters est un événement annuel de deux jours qui réunit des professionnels de santé du monde entier prenant en charge des patients atteints d’hypophosphatémie liée à l’X (XLH), toutes disciplines confondues. Lancée virtuellement en 2021, cette rencontre a lieu en présentiel depuis 2022 ; l’édition 2024 s’est tenue à Madrid les 18–19 avril et a rassemblé 122 participants issus de 27 pays, incluant des endocrinologues (pédiatriques et adultes), néphrologues, rhumatologues, chirurgiens orthopédistes, etc. Un compte-rendu détaillé de cette réunion a été publié par L. Seefried et al. en 2025, lequel constitue la source principale des données présentées ici.
Des études ont confirmé l’utilité du dosage de FGF23 comme marqueur précoce de la maladie, avec des taux significativement élevés chez les enfants XLH. Le traitement par burosumab, un anticorps anti-FGF23, s’est montré efficace à long terme sur le rachitisme, la croissance et la douleur, y compris chez des enfants ayant commencé par un traitement conventionnel. Par ailleurs, des données récentes suggèrent une amélioration de la taille adulte en France depuis 2001, attribuable à des diagnostics plus précoces et à l’accès au burosumab.
L’XLH affecte aussi les muscles et les tendons : les enfants et adultes XLH ont des muscles plus courts et moins puissants, avec une bioénergétique altérée. Des études sur souris montrent que le burosumab améliore la structure dentaire, tandis que le rôle du calcitriol est confirmé dans la prévention des enthésopathies (calcifications des insertions tendineuses). Ces découvertes renforcent l’importance d’un traitement précoce et ciblé.
Expériences internationales : vers de meilleures pratiques
Trois centres (Italie, Royaume-Uni, Bahreïn) ont partagé leurs ajustements après XLH Matters :
- Meilleur suivi de la croissance segmentaire grâce à la mesure de la taille assise et de l’envergure ;
- Renforcement du dépistage dentaire systématique ;
- Diagnostic et mise sous burosumab d’un adulte jusque-là non diagnostiqué, avec amélioration clinique notable.
Le vécu des patients : un pilier essentiel
Une analyse des réseaux sociaux a révélé les principaux besoins exprimés par les personnes vivant avec l’XLH :
- L’importance des groupes de soutien pour briser l’isolement ;
- Le besoin de communication fluide entre professionnels ;
- Le risque d’arrêt prématuré du traitement chez l’adulte, parfois vécu comme une « fausse guérison ».
Des efforts restent nécessaires pour garantir un accès équitable aux soins, notamment au burosumab à l’âge adulte, encore indisponible dans certains pays.
Pour en savoir + : XLH Matters 2024: expert insights and practical tools for enhancing care of people living with X-linked hypophosphataemia.
Seefried L, Alzahrani AS, Wagner CA, Eade D, Fintini D, Haffner D, Frookh Jamal H, Bubbear JS, Guazzarotti L, Cheung MS, Abid N, Costa-Reis P, Ferreira Santos R, Beck-Nielsen SS, Linglart A.Orphanet J Rare Dis. 2025 Sep 18;20(Suppl 2):477.
Cystinose : traitement oral de « nouvelle génération »
Les traitements usuels par cystéamine (Cystagon©, Procysbi©) prolongent la vie des patients, mais sont lourds (plusieurs prises quotidiennes) et entraînent des effets secondaires fréquents (odeur corporelle, troubles digestifs, ulcérations, etc.). Un cas récent (décrit par une équipe de Lyon, France) a par ailleurs mis en évidence une complication rare mais grave liée à la cystéamine à libération prolongée : une fillette de 10 ans atteinte de cystinose, traitée au long cours par cette formulation, a développé une colopathie fibreuse sévère avec sténose colique nécessitant une hémicolectomie. L’analyse histologique a révélé la présence de dépôts d’excipients (copolymères) dans la muqueuse digestive, suggérant un lien possible avec la formulation retard.
Fin 2025, la société Thiogenesis Therapeutics a annoncé le passage prochain en phase III de son candidat TTI-0102, un promédicament de la cystéamine conçu pour être pris une seule fois par jour.
Le TTI-0102 se présente comme une molécule combinant deux unités de cystéamine liées à de la pantothénate (vitamine B5). Après administration orale, cette prodrogue se métabolise lentement en libérant la cystéamine de façon prolongée, ce qui permettrait de maintenir un taux sanguin efficace avec une seule dose quotidienne. Les études initiales indiquent également une réduction du pic plasmatique de cystéamine, donc une meilleure tolérance gastro-intestinale par rapport aux formulations actuelles. Le développement bénéficie de l’expérience acquise sur la cystéamine retard (Procysbi©) et utilise la voie réglementaire de la FDA (référence à un produit existant) pour accélérer l’évaluation. Si les essais cliniques, confirment l’efficacité équivalente avec moins d’effets indésirables, le TTI-0102 pourrait représenter un progrès majeur en qualité de vie pour plus de 2 000 patients dans le monde en améliorant l’adhésion thérapeutique.
Vers un consensus européen sur la biopsie osseuse dans la MRC : une technique à revaloriser, y compris pour les maladies rares ?
Dans cet article de consensus publié en 2025 dans Bone, un groupe d’experts issus de 14 pays européens propose pour la première fois un cadre harmonisé pour la réalisation et l’interprétation de la biopsie osseuse iliaque dans le contexte de l’ostéodystrophie rénale (renal osteodystrophy, ROD). Cette démarche vise à redonner toute sa place à la biopsie osseuse histomorphométrique, considérée comme le gold standard dans l’évaluation du tissu osseux chez les patients atteints de maladie rénale chronique (MRC), mais dont l’usage clinique a fortement diminué ces dernières années.
Le document décrit de manière détaillée les bonnes pratiques à adopter tout au long du processus : indication clinique, technique de prélèvement, préparation des lames, paramètres recommandés, interprétation selon la classification TMV (Turnover, Mineralization, Volume), et rédaction du compte rendu. L'ensemble des recommandations a été validé par méthode Delphi, avec un seuil de consensus de 70 %.
Les auteurs insistent sur l’importance de la biopsie osseuse dans certaines situations où les outils standards, tels que la densitométrie osseuse (DXA), les marqueurs de remodelage ou même les imageries avancées – ne permettent pas de caractériser correctement l’état osseux. C’est notamment le cas lorsqu’il s’agit de différencier une ostéoporose classique d’une ostéopathie métabolique ou d’une anomalie de minéralisation. Or, c’est précisément dans ces contextes que se situent de nombreuses maladies osseuses rares.
Ainsi, bien que les maladies rares ne soient pas au cœur de l’article, leur mention implicite traverse plusieurs recommandations. La biopsie est présentée comme un outil indispensable dans les cas complexes ou atypiques, où les diagnostics différentiels incluent des pathologies rares comme l’hypophosphatasie, l’ostéomalacie héréditaire, les anomalies de minéralisation, certaines formes d’ostéopétrose, ou encore les ostéopathies liées à des troubles du métabolisme phosphocalcique. Dans ces cas, l’analyse histomorphométrique permet non seulement de quantifier le volume osseux et l’activité de remodelage, mais surtout d’observer directement les défauts de minéralisation ou les excès d’ostéoïde, éléments souvent cruciaux pour poser un diagnostic.
L’article souligne aussi l’utilité de la biopsie osseuse en pédiatrie, dans les formes précoces de maladies osseuses rares, en adaptant la technique de prélèvement à l’anatomie de l’enfant. Il appelle à la constitution de bases de données de références spécifiques à l’âge et au sexe, encore largement absentes aujourd’hui, ce qui constitue un obstacle majeur au diagnostic des maladies rares dans la population pédiatrique.
Enfin, les auteurs recommandent que les centres spécialisés s’organisent en réseau, notamment en Europe, pour mutualiser les données, échanger les lames digitalisées, comparer les interprétations, et former une nouvelle génération de spécialistes en histomorphométrie osseuse. Cette dynamique est essentielle si l’on souhaite faire de la biopsie osseuse un outil clinique réellement accessible, notamment dans les situations les plus complexes, comme celles rencontrées dans les centres de référence maladies rares.
Pour en savoir + : Bone histomorphometry for the diagnosis of renal osteodystrophy - a European consensus statement.
Lafage-Proust MH, Jørgensen HS, Bravenboer N, Ferreira A, Bégin MJ, Cannata-Andia J, Cejka D, Chavassieux P, Cohen-Solal M, D'Haese P, Fahrleitner-Pammer A, Ferreira AC, Fusaro M, Gerbaix M, Hamdy N, Hansen D, de Jongh R, Kröger H, Lalayiannis AD, Salam S, Spasovski G, Shroff R, Tong X, Trombetti A, Ureña P, Bacchetta J, Mazzaferro S, Haarhaus M, Evenepoel P; European Renal Osteodystrophy initiative of the CKD-MBD working group of the European Renal Association.Bone. 2025 Oct;199:117544.


