Actu MOC - Octobre 2025

Sommaire
🔎 Achondroplasie : études CANOPY
🔎 Un métabolite de “sauvetage” enzymatique manquant à l’origine d’une dysplasie squelettique rare
🔎 Syndrome de Miller : élargissement du spectre phénotypique dans une cohorte française
🔎 Étude TOPAZ : tériparatide→zolédronate versus soins usuels dans l’OI
🔎 AGA2115 (bispécifique sclérotine/DKK1) dans l’OI
🔎 OI: un double fardeau pour les aidants ?
🔎 Ostéolyse carpo-tarsienne multicentrique (MCTO) : ce que montre le modèle murin MafB P59L
🔎 Défaut osseux critique : la membrane prépare, l’autogreffe consolide
🔎 Réparation osseuse : régulation transcriptomique et épigénétique des cellules souches/progénitrices squelettiques
Achondroplasie : études CANOPY
Lors de l’ASBMR (American Society for Bone and Mineral Research ) Meeting 2025, le laboratoire BioMarin a présenté de nouvelles données issues des études CANOPY évaluant le vosoritide (VOXZOGO®) chez des enfants et adolescents atteints d’achondroplasie, avec un focus sur la morphologie rachidienne et la croissance post-pubertaire.
Phase 2 – CANOPY 111-206 (enfants ≤ 5 ans)
Cette étude randomisée contrôlée (n=67) visait à explorer l’impact de vosoritide sur la morphologie spinale, en particulier en lien avec la sténose du canal rachidien lombaire, une complication redoutée.
Après 52 semaines de traitement :
- Amélioration significative de l’interpedicular distance (IPD) et de la largeur du canal rachidien lombaire (L1–L5)
- Réduction de la cyphose thoraco-lombaire (TLK) chez 57 % des enfants sous vosoritide, contre 33 % sous placebo
Ces résultats suggèrent que vosoritide pourrait contribuer à prévenir les complications rachidiennes précoces telles que la sténose spinale.
Phase 3 – CANOPY 111-302 (extension post-pubertaire)
Chez les adolescents traités jusqu’à la fermeture des cartilages de croissance :
- Garçons : +7,55 cm de croissance (24,6 vs 17,1 cm) entre le début de la puberté (moy. 12,1 ans) et 18 ans
- Filles : +8,07 cm de croissance (21,2 vs 13,1 cm) entre 10,7 ans et 16 ans
Ces données renforcent la stratégie de traitement prolongé par vosoritide après la puberté, tant que les plaques de croissance restent ouvertes.
En résumé, vosoritide, au-delà de son effet sur la taille, montre un impact morphologique vertébral potentiellement protecteur et prolonge la croissance après la puberté. Ces résultats étendent significativement son champ de bénéfice chez les patients atteints d’achondroplasie.
Un métabolite de “sauvetage” enzymatique manquant à l’origine d’une dysplasie squelettique rare
Cette étude de Nature lève enfin le voile sur TGDS : l’enzyme est une UDP-glucose-4,6-déshydratase qui fabrique l’UDP-4-keto-6-désoxyglucose, un « métabolite de secours » capable de réactiver UXS1 en réoxydant le NADH piégé dans son site actif. Or, dans le RE/Golgi, l’enzyme H6PD maintient un NAD⁺ faible ; sans TGDS, UXS1 s’éteint et la production d’UDP-xylose chute, perturbant la synthèse des GAG et la glycosylation d’α-dystroglycane. Ces défauts sont corrigés par la ré-expression de TGDS, par des orthologues bactériens/fongiques qui fournissent le même sucre 4-céto, ou par la sur-expression d’UXS1 ; inversement, moduler H6PD rend la dépendance à TGDS plus ou moins marquée selon le type cellulaire. Un modèle murin Tgds^A100S/− retrouve un raccourcissement des os longs et des altérations crânio-faciales, avec baisse du métabolite de secours et déséquilibre UDP-xylose/UDP-glucuronate ; plusieurs variants TGDS humains montrent une activité enzymatique diminuée.
Cette étude bascule TGDS du statut « enzyme orpheline » à celui de gardien métabolique d’UXS1, et explique le lien entre variants TGDS et dysplasie squelettique via un défaut de GAG. Elle apporte : (i) un cadre mécanistique utile à l’interprétation des variants (diagnostic moléculaire, conseil génétique) ; (ii) des pistes de biomarqueurs (suivis ciblés des UDP-4-keto-6-désoxyglucose/UDP-6-désoxyhexose dans des matrices appropriées) ; (iii) des leviers thérapeutiques conceptuels à explorer : fourniture de métabolites 4-céto-sucres « secours », modulation de l’axe H6PD–NAD⁺/NADH dans le RE/Golgi, ou augmentation d’UXS1. À ce stade, il s’agit de pistes précliniques (pas de traitement prêt-à-l’emploi) mais la démonstration que des « métabolites de sauvetage » peuvent réactiver des enzymes ouvre une classe d’approches transposables à d’autres maladies métaboliques rares et communes.
Pour en savoir + : A missing enzyme-rescue metabolite as cause of a rare skeletal dysplasia.
Jacobs J, Lyubenova H, Potelle S, Kopp J, Gerin I, Chan WL, Rodriguez de Los Santos M, Hülsemann W, Mensah MA, Cormier-Daire V, Joosten M, Bruggenwirth HT, Stuurman KE, Miranda V, Campeau PM, Wittler L, Graff J, Mundlos S, Ibrahim DM, Van Schaftingen E, Fischer-Zirnsak B, Kornak U, Ehmke N, Bommer GT.Nature. 2025 Oct;646(8083):218-226.
Syndrome de Miller : élargissement du spectre phénotypique dans une cohorte française
Dans cette étude multicentrique française, les auteurs décrivent 10 personnes issues de 7 familles atteintes du syndrome de Miller (acro-facio-dysostose liée à des variants bialléliques de DHODH), de la période prénatale jusqu’à 46 ans, constituant à ce jour la plus large cohorte publiée.
Le tableau confirme les atteintes post-axiales des membres (absence du 5ᵉ rayon mains/pieds) mais documente aussi, plus fréquemment qu’attendu, des atteintes pré-axiales (hypoplasie du pouce et du tibia), avec camptodactylie et synostoses radio-cubitales fréquentes ; les traits crânio-faciaux associent rétrognathie/micrognathie et fentes (labiale/palatine ou luette bifide), tandis que les cardiopathies congénitales (surtout CIV auriculaire) sont récurrentes.
Fait rassurant pour le suivi : tous les sujets vivants ont un neurodéveloppement normal. L’étude signale deux éléments nouveaux d’intérêt clinique : la présence d’un naevus simplex facial chez deux enfants et l’atrophie optique regroupée dans une famille consanguine, observation qui appelle un dépistage ophtalmologique ciblé dans les formes confirmées. Sur le plan moléculaire, seuls des variants faux-sens de DHODH sont rapportés, touchant le domaine catalytique, avec des situations homozygotes (y compris un pseudo-dominant familial) et composés hétérozygotes ; aucun hotspot ni corrélation génotype-phénotype n’émerge.
Pour la pratique, l’association « défauts des rayons post-axiaux ± pré-axiaux + synostoses + cardiopathie » doit faire évoquer le diagnostic dès l’échographie prénatale et conduire à un séquençage DHODH ; en postnatal, penser à un bilan cardiaque systématique, à une évaluation visuelle et auditive régulière, et à la prise en charge chirurgicale des fentes et des malformations des extrémités selon besoin. Les limites tiennent à la taille d’échantillon et au recueil rétrospectif, mais cette série élargit nettement le spectre phénotypique et permet de clarifier la prise en charge initiale.
Pour en savoir + : The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
Aubert-Mucca M. and al. Clinical Genetics, 2025; 0:1–6 https://doi.org/10.1111/cge.70015
Étude TOPAZ : tériparatide→zolédronate versus soins usuels dans l’OI
Lors de l’ASBMR (American Society for Bone and Mineral Research ) Meeting 2025, l’équipe du projet TOPAZ a présenté des résultats initiaux d’un essai randomisé chez 349 adultes atteints d’ostéogenèse imparfaite (OI). Les participants ont été tirés au sort pour recevoir soit un traitement anabolique puis anti-résorptive (deux ans de tériparatide (TPTD, médicament qui stimule la formation d’os) suivis d’une perfusion unique de zolédronate (ZA, bisphosphonate qui freine la perte d’os), soit les soins habituels décidés par leur médecin, où l’usage de bisphosphonates était possible mais sans médicament anabolique. L’objectif principal était de voir si la stratégie tériparatide→zolédronate réduisait le nombre de fractures cliniques confirmées par imagerie, par rapport aux soins habituels, sur un suivi médian d’environ 4 ½ ans.
Le critère primaire (proportion de sujets avec fracture clinique confirmée par imagerie et adjudiquée en aveugle) n’est pas différent entre groupes : 36,9 % sous TPTD→ZA versus 35,8 % sous soins habituels. En revanche, la densité minérale osseuse (DMO) vertébrale augmente significativement et durablement avec TPTD→ZA, tandis que le col fémoral/hanche totale progresse sans atteindre la significativité inter-groupes ; les marqueurs de remodelage sont plus élevés sous TPTD→ZA à toutes les échéances. La cohorte (âge moyen 43,7 ans ; 54 % de femmes) compte majoritairement des OI type I (76 %) avec variants COL1A1/COL1A2 (88 %) ; suivi médian 55,5 mois, 86,5 % complétés.
Malgré un gain de DMO vertébrale et une stimulation du remodelage, la séquence TPTD→ZA ne réduit pas les fractures cliniques par rapport aux soins habituels dans cette analyse initiale ; des analyses en cours porteront sur les fractures totales et vertébrales morphométriques.
Pour en savoir + : 1127 - Initial results of the TOPAZ study: A randomised trial of teriparatide and zoledronic acid compared with standard care in adults with osteogenesis imperfecta. Stuart H. Ralston and al. ASBMR Meeting 2025.
AGA2115 (bispécifique sclérotine/DKK1) dans l’OI
Une avancée majeure présentée lors de l’ASBMR (American Society for Bone and Mineral Research ) Meeting 2025 est AGA2115, un anticorps bispécifique ciblant simultanément la sclérotine et la DKK1 (deux inhibiteurs clés de la voie Wnt dans l’os). Lors d’un essai de Phase 1 (chez des volontaires sains), l’administration d’AGA2115 a entraîné des augmentations dose-dépendantes de la densité minérale osseuse, DMO (jusqu’à +14,4 % de DMO lombaire en 6 mois à la dose élevée). Ces augmentations de DMO étaient accompagnées d’une élévation rapide et soutenue des marqueurs de formation osseuse (par exemple ostéocalcine, P1NP) et d’une diminution des marqueurs de résorption, confirmant un effet anabolique marqué.
Le profil de tolérance était favorable à tous les niveaux de dose. Les effets indésirables enregistrés étaient peu fréquents et comparables au placebo, et aucun effet indésirable grave lié au traitement n’a été rapporté.
Des données précliniques associées ont montré qu’un anticorps bispécifique analogue (anti-DKK1/anti-sclérotine) réduisait le nombre de fractures spontanées et améliorait la qualité osseuse chez des souris modèles d’OI.
Sur la base de ces résultats, le laboratoire pharmaceutique prévoit de lancer un essai clinique de phase 2 chez des patients adultes atteints d’OI. Une extension aux populations pédiatriques est envisagée par la suite. AGA2115 bénéficie par ailleurs de désignations de médicament orphelin (FDA et EMA), reconnaissant le besoin médical non satisfait dans l’OI.
OI : un double fardeau pour les aidants ?
Deux volets complémentaires de l’IMPACT Survey dressent un tableau cohérent du vécu des 528 aidants sans ostéogenèse imparfaite (OI) s’occupant chacun d’une personne atteinte : un coût économique tangible et un retentissement humain marqué. Côté économie, parmi les aidants en emploi (64 %), 1 sur 2 a manqué au moins une journée de travail au cours des 4 semaines précédentes (moyenne 1,9 jour) ; les dépenses directes atteignent en moyenne 209 €/4 semaines, surtout pour les déplacements médicaux (83 €) et les médicaments (46 €), avec de fortes variations régionales (USA 334 € vs 163 € en EU+UK et 33 € dans les pays nordiques). Des facteurs liés à l’aidant (âge, statut professionnel) et au proche (âge, sévérité, signes/événements cliniques) modulent ces impacts ; par exemple, des problèmes dentaires ou de poids bas chez le proche augmentent nettement la probabilité d’absences professionnelles.
Sur le plan « humaniste », 58-83 % des aidants rapportent une atteinte de la qualité de vie, avec une pression particulière sur la santé mentale (≈ 80 %) et le temps libre (≈ 83 %) ; les inquiétudes sont quasi unanimes pour l’avenir, l’accès aux traitements et la transition pédiatrie-adulte. Là encore, l’âge de l’aidant, la sévérité de l’OI et des événements cliniques chez le proche sont associés à un retentissement plus important. Pris ensemble, ces résultats plaident pour des dispositifs de soutien ciblés (santé mentale, répit, aide aux déplacements), une anticipation de la transition de soins et des aménagements professionnels pour limiter la perte de productivité.
Pour en savoir + : The IMPACT Survey: The Humanistic Impact of Caring for an Individual with Osteogenesis Imperfecta.
Westerheim I, Rauch F, Hart T, Wekre LL, van Welzenis T, Raggio C, Mulhall H, Battersby A, Prince S, Semler O.Adv Ther. 2025 Sep 26. doi: 10.1007/s12325-025-03372-8. Online ahead of print.PMID: 41004075
The IMPACT Survey: The Economic Impact of Caring for an Individual with Osteogenesis Imperfecta.
Rauch F, van Welzenis T, Wekre LL, Raggio C, Semler O, Westerheim I, Mulhall H, Anderson M, Prince S, Hart T.Adv Ther. 2025 Sep 23. doi: 10.1007/s12325-025-03373-7. Bas du formulaire
Ostéolyse carpo-tarsienne multicentrique (MCTO) : ce que montre le modèle murin MafB P59L
La MCTO, ultra-rare, est due à des variants faux-sens hétérozygotes dans le domaine d’activation de MafB et se traduit chez l’humain par une ostéolyse progressive des os du carpe et du tarse. Pour en explorer les mécanismes, les auteurs ont créé une souris knock-in MafB P59L (hétérozygote KI/+ et homozygote KI/KI).
À 4, 8 et 12 semaines, la micro-CT montre une baisse dose-dépendante du volume osseux au tibia, fémur et vertèbres. Paradoxalement, l’histomorphométrie et le P1NP sérique (marqueur de formation) augmentent avec la dose génique, suggérant un remodelage accéléré « formation ↑ mais insuffisante pour compenser la résorption ». In vivo, le nombre d’ostéoclastes est plus élevé, mais les marqueurs circulants CTX (résorption) ne changent pas et TRAP5b diminue, témoignant d’un décalage entre activité locale et signal sanguin.
Invitro, les progéniteurs stromaux médullaires mutants (KI/KI) forment moins de colonies (fibroblastiques/ostéoblastiques : CFU-F, CFU-OB) et les ostéoblastes (OB) calvariens se différencient mal. Côté lignée myéloïde, les macrophages médullaires (BMM) KI/KI forment moins d’ostéoclastes (OC) sous M-CSF/RANKL, et les co-cultures BMM–ostéoblastes révèlent que les deux compartiments (OB et BMM) soutiennent moins bien l’ostéoclastogenèse. Autrement dit, la mutation altère intrinsèquement les deux lignées osseuses.
L’immunohistochimie détecte MafB dans plusieurs tissus, notamment le cartilage articulaire. Les western blots montrent une augmentation de MafB dans les chondrocytes, les ostéoblastes et les BMM mutants ; l’exposition des BMM à RANKL abaisse toutefois MafB. Les analyses transcriptomiques (RNA-seq) menées sur l’os cortical, le cartilage, la synoviale, les ostéoblastes, les BMM et les ostéoclastes révèlent des modulations dose-dépendantes, avec hausse de facteurs pro-résorptifs (dont RANKL) et renforcement d’interactions inflammatoires de bas bruit dans tous les tissus mutants. Phénotypiquement, les souris ne présentent pas une « disparition » complète des os carpiens/tarsiens, mais montrent des érosions sous-chondrales ostéoclastiques et des ostéophytes d’allure arthrosique.
Ces données soutiennent l’exploration de stratégies ciblant l’axe RANKL–RANK–OPG et l'inflammation locale de bas grade (cartilage/synoviale), au-delà des approches classiques purement anti-résorptives, et justifient des analyses monocellulaires (scRNA-seq) pour identifier les facteurs clés modulés par MafB P59L.
Pour en savoir + : 277 - Systemic Bone Loss, Periarticular Bone Resorption and Transcriptomic Changes in the Mafb P59L Mouse Model of Multicentric Carpo-Tarsal Osteolysis (MCTO). Byeong-Rak Keum and al. ASBMR Meeting 2025.
Défaut osseux critique : la membrane prépare, l’autogreffe consolide
L’article présente un modèle préclinique pour étudier la technique de membrane induite de Masquelet chez le rat, avec un objectif très concret : savoir dans quelles conditions un défaut osseux critique peut réellement consolider et si un biomatériau peut remplacer la greffe. La technique, rappelons-le, se fait en deux temps : pose d’un espaceur en PMMA qui induit une membrane biologique, puis, 4 à 6 semaines plus tard, retrait du PMMA et comblement par autogreffe, la membrane jouant le rôle de « chambre de régénération ». Les auteurs rappellent que cette approche est efficace en clinique mais exige souvent de grandes quantités d’os autologue ; d’où l’intérêt de tester des substituts pour économiser l’autogreffe.
Le modèle mis au point est un défaut fémoral de 4 mm chez le rat Lewis, stabilisé par plaque vissée et suivi jusqu’à 24 semaines en micro-CT et en histologie. Le travail comporte d’abord une phase d’optimisation (comment couper l’os et comment fixer), puis une phase de validation comparant plusieurs scénarios : défaut laissé vide, chirurgie en un temps avec greffe syngénique, chirurgie en deux temps avec membrane seule, deux temps avec greffe, et deux temps avec verre bioactif (GlassBone™) comme substitut.
L’optimisation est très instructive pour la pratique : la piezotomie s’avère la meilleure ostéotomie (coupes nettes, tissus respectés, canaux haversiens « perméables »), tandis que scie ou fraise entraînent brûlures des berges et fragilisation. Côté matériel, seule la plaque 1,0 mm avec vis 2,0 mm reste en place ; la plaque fine (0,6 mm) échoue systématiquement à 6 semaines. Autrement dit, stabilité rigide et coupes atraumatiques conditionnent la viabilité du modèle, un message transposable à la chirurgie humaine.
Vient le résultat central : à 24 semaines, la consolidation complète n’est obtenue que lorsque l’on réalise les deux temps ET que l’on greffe (groupe 2T-S). La membrane seule (2T-Empty) ne ferme pas le défaut ; la chirurgie en un temps ne le fait pas non plus. Avec GlassBone™ utilisé seul, on observe des îlots d’ossification au contact du biomatériau, mais pas de pontage osseux complet. En revanche, la condition « deux temps + verre » préserve mieux le matériau et réduit le tissu cicatriciel, ce qui suggère que la membrane joue son rôle de contenant vascularisé, mais insuffisant sans apport d’os autologue. Le choix d’un suivi long (24 semaines) est volontaire : plusieurs modèles publiés s’arrêtent à 8-12 semaines, délai trop court pour vérifier la corticalisation complète.
Ce modèle valide que 4 mm est bien un défaut critique dans ces conditions et qu’il est pertinent pour évaluer des biomatériaux en situation Masquelet. Mais il confirme surtout que, en l’absence d’autogreffe, ni la membrane ni un substitut utilisé seul ne rétablit l’intégrité mécanique du segment osseux.
Les auteurs proposent donc logiquement la suite : tester des combinaisons greffe + biomatériau pour épargner du greffon. Ils rappellent d’ailleurs que, en clinique rachidienne, le verre bioactif combiné à l’autogreffe donne des résultats comparables à l’autogreffe seule ; c’est compatible avec l’idée d’un rôle adjuvant du matériau, plutôt que substitutif.
Ce travail renforce la doctrine des équipes : dans les défauts critiques, la stratégie la plus fiable reste la procédure en deux temps avec autogreffe.
Pour en savoir + : Optimisation and Validation of an Induced Membrane Technique Model to Assess Bone Regeneration in Rats.
Siboni R, Sergheraert J, Thoraval L, Guillaume C, Gangloff SC, Ohl X, Braux J, Velard F. J Tissue Eng Regen Med. 2025 Apr 21;2025:7357277. doi: 10.1155/term/7357277. eCollection 2025.
Réparation osseuse : régulation transcriptomique et épigénétique des cellules souches/progénitrices squelettiques
Cette étude montre que, lors de la réparation d’une fracture, les cellules souches/progénitrices du squelette (SSPCs) ne contribuent pas toutes de la même façon selon leur origine tissulaire. En traçant génétiquement les lignées cellulaires chez la souris, les auteurs établissent que les SSPCs du périoste et des muscles adjacents sont les principales sources de nouveaux tissux osseux, alors que les SSPCs médullaires (moelle) apportent une contribution minime à l’ossification intramembraneuse.
La déplétion ciblée des SSPCs périostées/musculaires empêche la consolidation, confirmant qu’elles sont indispensables à la guérison. Les deux populations (périoste et muscle) participent à l’ossification endochondrale (passage par un cartilage transitoire), mais seules les SSPCs périostées réalisent en plus une ossification intramembraneuse directe (os formé sans étape cartilagineuse).
Sur le plan moléculaire, l’expression de SOX9 (facteur clé du chondrogène) diverge entre ces compartiments : inactiver SOX9 bloque l’ossification endochondrale mais n’affecte pas l’ossification intramembraneuse issue du périoste. Des analyses multiomiques (ARN nucléaire + ATAC-seq) au repos indiquent d’ailleurs que les SSPCs périostées sont pré-équipées épigénétiquement pour l’ossification intramembraneuse, tandis que périoste et muscle partagent un programme commun pour l’endochondrale.
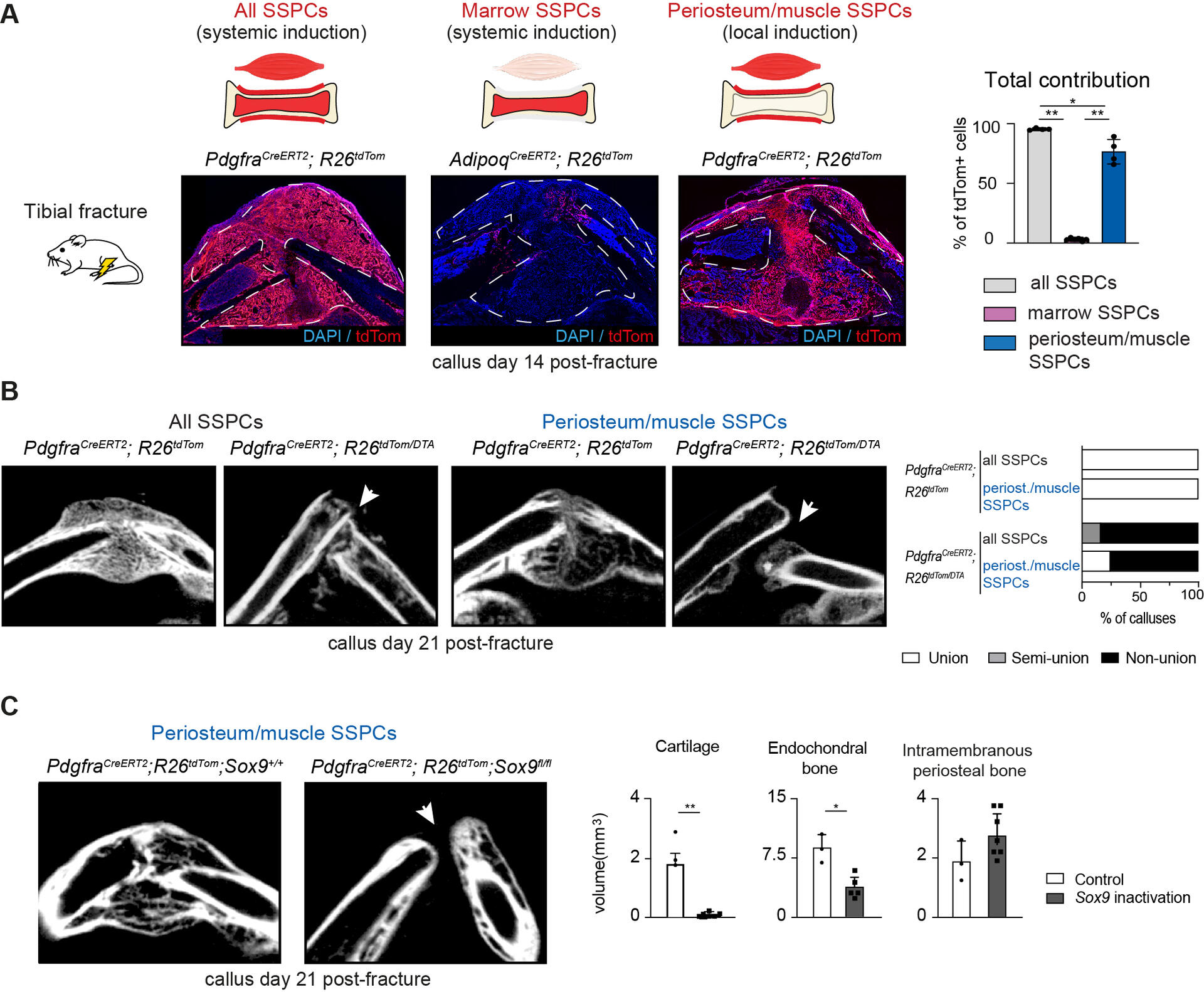
Sur la figure A, le marquage (Pdgfra^CreERT2 ou AdipoQ^CreERT2) révèle que, 14 jours après fracture, la grande majorité des cellules du cal mobilisées provient du périoste/muscle, pas de la moelle. En B, l’imagerie à J21 montre qu’éliminer ces SSPCs périostées/musculaires conduit à des non-consolidations, soulignant leur rôle essentiel. En C, l’inactivation de SOX9 dans les SSPCs périostées réduit nettement le cartilage et l’os endochondrale, mais épargne l’os intramembraneux formé au périoste, ce qui matérialise la dépendance à SOX9 de la voie endochondrale uniquement.
En pratique, ces données affinent notre compréhension de la biologie de la consolidation : le périoste est le réservoir prioritaire à cibler/stimuler, notamment pour l’ossification directe, tandis que la voie chondrogène dépendante de SOX9 demeure cruciale pour la composante endochondrale. Cela ouvre des pistes pour des stratégies de thérapie cellulaire ou de guidage moléculaire différenciées selon l’origine des SSPCs et le type d’ossification recherché.
Pour en savoir + : 1094 - Transcriptomic and epigenetic regulation of Skeletal stem/progenitor cells during bone regeneration. Maria Ethel and al. ASBMR Meeting 2025.


