Actu CAP - Octobre 2025

Sommaire
🔎 Hypophosphatémie liée à l’X : fiabiliser la stadification rénale au-delà de la créatinine
🔎 Hypophosphatasie : des porteurs ALPL “silencieux” à phénotype biochimique
🔎 Qualité de vie dans l'XLH pédiatrique française : signaux favorables sous burosumab
🔎 FGF23 et anémie par carence martiale : ce n’est pas l’os, c’est l’érythroblaste !
Hypophosphatémie liée à l’X : fiabiliser la stadification rénale au-delà de la créatinine
L’étude, menée à Lyon dans un centre expert, part d’un constat clinique : chez l’adulte XLH, les formules d’estimation de la fonction rénale qui utilisent la créatinine surestiment souvent le débit de filtration glomérulaire (DFG), car la créatinine dépend de la masse musculaire, fréquemment diminuée dans cette maladie. Les auteurs comparent donc, chez des adultes XLH, les DFG estimés par CKD-EPI (créatinine et cystatine C) au DFG mesuré par clairance à l’iohexol, et explorent l’impact potentiel du FGF23 (et du traitement par burosumab) sur la filtration glomérulaire.
Il s’agit d’une étude rétrospective, monocentrique, incluant 20 adultes XLH (17 femmes, âge médian 35 ans). Pour discuter le rôle hémodynamique du FGF23, l’équipe a aussi analysé la relation FGF23–eGFR dans une cohorte pédiatrique indépendante atteinte de dysplasie fibreuse (autre maladie rare avec FGF23 élevé).
Les résultats principaux confirment la surestimation par la créatinine. Le DGF mesuré médian est de 98 mL/min/1,73 m² (IQR 85–107), alors que le DFGe CKD-EPI basé sur la créatinine est de 125 (114–131) et celui basé sur la cystatine C de 111 (104–119). Le biais DFGm-DFGe est nettement plus défavorable avec la créatinine (−22 mL/min/1,73 m²) qu’avec la cystatine C (−13 mL/min/1,73 m²). L’exactitude (P30) est de 25 % pour CKD-EPIcreat contre 93 % pour CKD-EPIcyst. Concrètement, 5 patients sont mal classés en stades de maladie rénale chronique par la formule créatinine ; l’hyperfiltration est étiquetée chez 60 % des patients avec DGFe-créatinine, contre 10 % avec DFGe- cystatine C et 5 % seulement avec le DFGm.
Le profil rénal de la cohorte est rassurant sur certains points mais alerte sur d’autres : aucune néphrocalcinose à l’échographie ; micro-albuminurie chez 15 % ; hypertension chez 10 % ; hyperparathyroïdie secondaire chez 42 %.
Dans un sous-groupe de 5 adultes évalués avant et après le passage au burosumab, le DFGm diminue de 98 à 84 mL/min/1,73 m² (tendance, P = 0,06), soit un déclin annuel médian d’environ −4,2 mL/min/1,73 m². Les auteurs insistent sur la prudence d’interprétation (faible effectif), mais discutent deux mécanismes : d’une part, l’amélioration de la masse musculaire sous burosumab peut augmenter la créatinine et donc abaisser artificiellement l’DFGe créatinine ; d’autre part, le FGF23 pourrait influencer la dynamique de filtration via la balance sodée et l’activation du RAAS, de sorte qu’une baisse du DFG après anti-FGF23 pourrait refléter la normalisation d’une hyperfiltration préalable plutôt qu’une dégradation de la fonction rénale.
Pour étayer ce point, la cohorte pédiatrique en dysplasie fibreuse (n = 14, âge médian 12 ans) montre une corrélation significative entre FGF23 et DFGe suggérant un effet hémodynamique du FGF23 sur la filtration glomérulaire. Ces données, encore exploratoires, renforcent l’idée qu’il faut relire les trajectoires de DFG sous burosumab à la lumière de la physiologie du FGF23.
En conclusion, chez l’adulte XLH, la créatinine surestime la fonction rénale et expose à des erreurs de stadification ; la cystatine C fournit une estimation nettement plus fiable mais pas parfaite. Il est recommandé de mesurer la cystatine C au suivi courant, et de recourir à une mesure de DFG par iohexol lorsqu’il existe un enjeu clinique (suspicion de maladie rénale chronique, décision thérapeutique, évolution atypique ou sous burosumab). Un suivi systématique de la pression artérielle et de l’albuminurie est également préconisé pour détecter précocement une atteinte rénale discrète. Enfin, les variations du DFG observées sous anti-FGF23 doivent être interprétées avec prudence, en intégrant l’effet potentiel du FGF23 sur l’hémodynamique glomérulaire.
Pour en savoir + : Challenges in Estimating Renal Function in X-linked Hypophosphatemia because of formula Overestimation and FGF23 Effects.
Lemoine S, De Mul A, Perge K, Derain-Dubourg L, Chapurlat R, Vignot E, Legrand MA, Bacchetta J.J Clin Endocrinol Metab. 2025 Aug 25:dgaf459. doi: 10.1210/clinem/dgaf459. Online ahead of print.
Hypophosphatasie : des porteurs ALPL “silencieux” à phénotype biochimique
Cette étude du Journal of Bone and Mineral Research documente, à partir de la base mondiale des variants ALPL, un phénotype jusque-là mal défini : des porteurs de variants ALPL présentant un profil biochimique d’hypophosphatasie (HPP) (ALP sérique basse avec parfois PLP et/ou PEA élevées, mais sans symptômes cliniques).
Au total, 43 individus répondant à ces critères ont été identifiés (médiane 29 ans), dont 79 % hétérozygotes et 21 % bi-alléliques (homozygotes ou composés). Les ALP étaient abaissées chez tous, le plus souvent modérément (≈ 76 % à moins de 50 % sous la limite inférieure de la normale), et 19/43 avaient un substrat élevé (PLP et/ou PEA). Trente génotypes distincts sont décrits, majoritairement faux-sens, souvent hors domaines fonctionnels critiques ; fait notable, cinq variants à effet dominant-négatif démontré in vitro ont été observés chez des sujets néanmoins asymptomatiques, soulignant la variabilité génotype-phénotype et l’incomplète pénétrance dans l’HPP. Un suivi limité (≈2,5 ans, trois sujets) montre la persistance du profil biochimique sans conversion clinique sur la période observée. Les auteurs proposent de nommer ce groupe « porteurs ALPL asymptomatiques avec phénotype biochimique d’HPP » et insistent sur les implications diagnostiques : affiner les seuils et critères pour éviter la sur-diagnose, rechercher les causes non génétiques d’ALP basse, réserver la thérapie enzymatique (asfotase alfa) aux formes symptomatiques avérées ; les données de sécurité étant absentes dans ces formes et un risque de calcifications ectopiques étant théorique sous doses standards.
Pour en savoir + : Biochemical phenotype of hypophosphatasia in asymptomatic individuals carrying ALPL variants.
Montero-Lopez R, Farman MR, Högler F, Rehder C, Malli T, Webersinke G, Rockman-Greenberg C, Dahir K, Martos-Moreno GÁ, Linglart A, Ozono K, Seefried L, Del Angel G, Nading EB, Huggins E, Rush ET, Tauer JT, Kishnani PS, Högler W.J Bone Miner Res. 2025 Sep 11:zjaf124.
Qualité de vie dans l'XLH pédiatrique française : signaux favorables sous burosumab
Cette analyse « vie réelle » du Registre International XLH menée en France décrit la qualité de vie liée à la santé (PedsQL) chez 96 enfants (âge moyen 8,1 ans), dont 82 % recevaient du burosumab au moment de l’évaluation. Les scores montrent un impact plus marqué sur la santé psychosociale que physique, avec une dégradation notable chez les 5–7 ans comparativement à des témoins sains, tandis que les adolescents se rapprochent des valeurs de référence.
Dans cette étude, un taux de phosphate un peu plus élevé dans le sang (dans la normale) allait de pair avec de meilleurs scores psychosociaux, et les enfants traités par burosumab avaient en moyenne de meilleures capacités physiques ; à l’inverse, les enfants sous phosphate/vitamine D ont des scores globaux inférieurs. Par rapport à d’autres maladies musculosquelettiques pédiatriques, les enfants avec XLH recevant du burosumab présentent des scores significativement meilleurs. Toutefois, ces liens doivent être confirmés par des approches longitudinales ajustées. À court terme, l’âge 5-7 ans apparaît comme une période vulnérable à cibler pour l’accompagnement (douleur, émotions, interruptions scolaires), alors que les adolescents tendent vers des valeurs proches des témoins.
Pour en savoir + : Health-related quality of life in French pediatric patients with X-linked hypophosphatemia: real-world data from the International XLH Registry.
Linglart A, Amouroux C, Gueorguieva I, Harambat J, Salles JP, Ertl DA, Sandilands K, Rylands A, Williams A, Ishii H, Bowden A, Varni JW, Bacchetta J.JBMR Plus. 2025 Aug 30;9(10):ziaf142.
FGF23 et anémie par carence martiale : ce n’est pas l’os, c’est l’érythroblaste !
Le FGF23 (Fibroblast Growth Factor 23 ) est une hormone osseuse produite par les ostéocytes qui régule le phosphate. Lors d’anémie ferriprive (AF) ou d’insuffisance rénale chronique (IRC), le FGF23 est également synthétisé par les érythroblastes (lignée rouge), en plus des ostéocytes.
Pour distinguer l’effet de chaque source sur l’anémie, les auteurs ont créé des souris où Fgf23 est soit supprimé, soit sur-exprimé dans l’os ou dans la lignée rouge, puis ont provoqué une AF par régime pauvre en fer. Ils ont analysé le fer, l’hémogramme et la maturation des précurseurs érythroïdes, et testé in vitro l’effet du FGF23 intact (iFGF23) sur des progéniteurs, avec ou sans blocage du récepteur FGFR1.
Résultat : en carence martiale, l’os et l’érythroïde produisent du FGF23, mais seule la source érythroïde explique l’anémie. Supprimer Fgf23 dans la lignée rouge corrige l’AF alors que le faire dans l’os ne change rien ; à l’inverse, sur-exprimer Fgf23 dans l’érythroïde induit une anémie, ce que ne provoque pas la sur-expression osseuse. En culture, l’iFGF23 freine la maturation des progéniteurs érythroïdes : il maintient des marqueurs précoces (Kit, Mecom ), abaisse des marqueurs tardifs (Tfrc, Hba-a1), s’accompagne d’une dysfonction mitochondriale, et son effet est dose-dépendant. Cet effet est aboli par un inhibiteur de FGFR1 (PD173074), montrant une action paracrine directe d’iFGF23 via FGFR1 sur les précurseurs.
En synthèse, dans l’AF (et possiblement en IRC), c’est l’iFGF23 produit par la lignée érythroïde, et non l’iFGF23 d’origine osseuse, qui ralentit l’érythropoïèse et contribue à l’anémie. Sur le plan translationnel, ces données (précliniques) suggèrent de viser l’axe FGFR1 dans la moelle ou de réduire spécifiquement l’iFGF23 érythroïde, plutôt qu’un blocage systémique du FGF23 qui perturberait l’équilibre phosphocalcique.
Pour en savoir + : 1030 - Intact FGF23 produced by erythroid cells is a paracrine inhibitor of erythropoiesis. Jane Joy Thomas and al. ASBMR Meeting 2025.
BAMBI, pivot de l’anabolisme en rééquilibrant TGF-β/Wnt
L’équipe s’intéresse à la façon dont le signal PTH (parathormone) stimule l’os quand il est administré par intermittence, un mode d’action classiquement anabolique. En analysant par RNA-seq des ostéocytes (cellules osseuses “chef d’orchestre” du remodelage) de souris ovariectomisées traitées par PTH, un gène ressort fortement induit : BAMBI (Bone morphogenetic protein and Activin Membrane-Bound Inhibitor ). BAMBI est un récepteur-leurre du TGF-β de type I : en “épongeant” la voie TGF-β, il baisse ce signal et favorise la voie Wnt, deux voies majeures qui orientent le comportement de l’ostéocyte et, in fine, la formation d’os. Des bases scRNA-seq confirment que BAMBI est surtout exprimé par l’ostéocyte.
Les auteurs vérifient ensuite l’expression et la fonction de BAMBI in vitro. Dans deux lignées ostéocytaires (OmGFP66, Ocy454), la PTH augmente BAMBI. À l’inverse, éteindre BAMBI (lignes “BAMBI-KD”) désorganise l’équilibre des voies : Sost (qui code la sclérostine, inhibiteur de Wnt) et Tnfsf11/RANKL (stimulateur de résorption par les ostéoclastes) montent, la β-caténine active (Wnt fonctionnelle) baisse, et pSmad2 (TGF-β activée) augmente. Sur le plan de la fonction cellulaire, les cellules sans BAMBI minéralisent moins, expriment moins Fgf23, perdent leur morphologie dendritique (moins de prolongements) et abaissent Sp7/Osterix et Osteocrine (deux gènes clés pour l’identité et les dendrites de l’ostéocyte).
Point crucial : la PTH change de visage quand BAMBI manque. Normalement, la PTH réprime Sost (ce qui “libère” la voie Wnt) et augmente Fgf23. Dans les cellules BAMBI-KD, la PTH n’éteint plus Sost (elle l’augmente au contraire) et perd sa capacité à stimuler Fgf23. En revanche, la branche PTH → RANKL reste intacte malgré l’absence de BAMBI. Autrement dit, les réponses de l’ostéocyte à la PTH se séparent en deux bras : un bras Wnt/anabolique (répression de Sost, induction de Fgf23, dendrites), dépendant de BAMBI, et un bras RANKL/résorptif, indépendant de BAMBI. Une RNA-seq globale après PTH confirme cette bascule : les cibles de Wnt (Sfrp4, Axin2, Dkk) sont abaissées sans BAMBI, tandis que les cibles de TGF-β/BMP (Id1-3) sont renforcées.
Les auteurs testent enfin in vivo l’importance de BAMBI dans l’ostéocyte en croisant des souris Dmp1-Cre (10 kb) avec des BAMBI^flox : la micro-CT montre une diminution significative de la masse osseuse (tibia, vertèbres) quand BAMBI est supprimé dans l’ostéocyte, ce qui relie directement ce gène à l’homéostasie squelettique.
Que faut-il retenir ? BAMBI est une cible majeure et nécessaire de la PTH dans l’ostéocyte. Il oriente l’aiguille entre TGF-β (plutôt frein) et Wnt (plutôt accélérateur anabolique), et il est indispensable pour que la PTH réprime Sost (sclérostine), stimule FGF23 et entretienne l’architecture en dendrites des ostéocytes, autant d’éléments qui participent à l’effet constructeur d’os. En revanche, la production de RANKL par la PTH ne dépend pas de BAMBI, ce qui dissocie deux voies de signalisation et suggère qu’il pourrait être possible, à terme, de renforcer sélectivement le bras anabolique PTH/Wnt sans amplifier le bras RANKL. Il s’agit d’un travail préclinique, mais il éclaire finement la biologie de l’ostéocyte et ouvre des pistes pour optimiser les thérapies anaboliques de l’os en ciblant le couple PTH–BAMBI–Wnt.
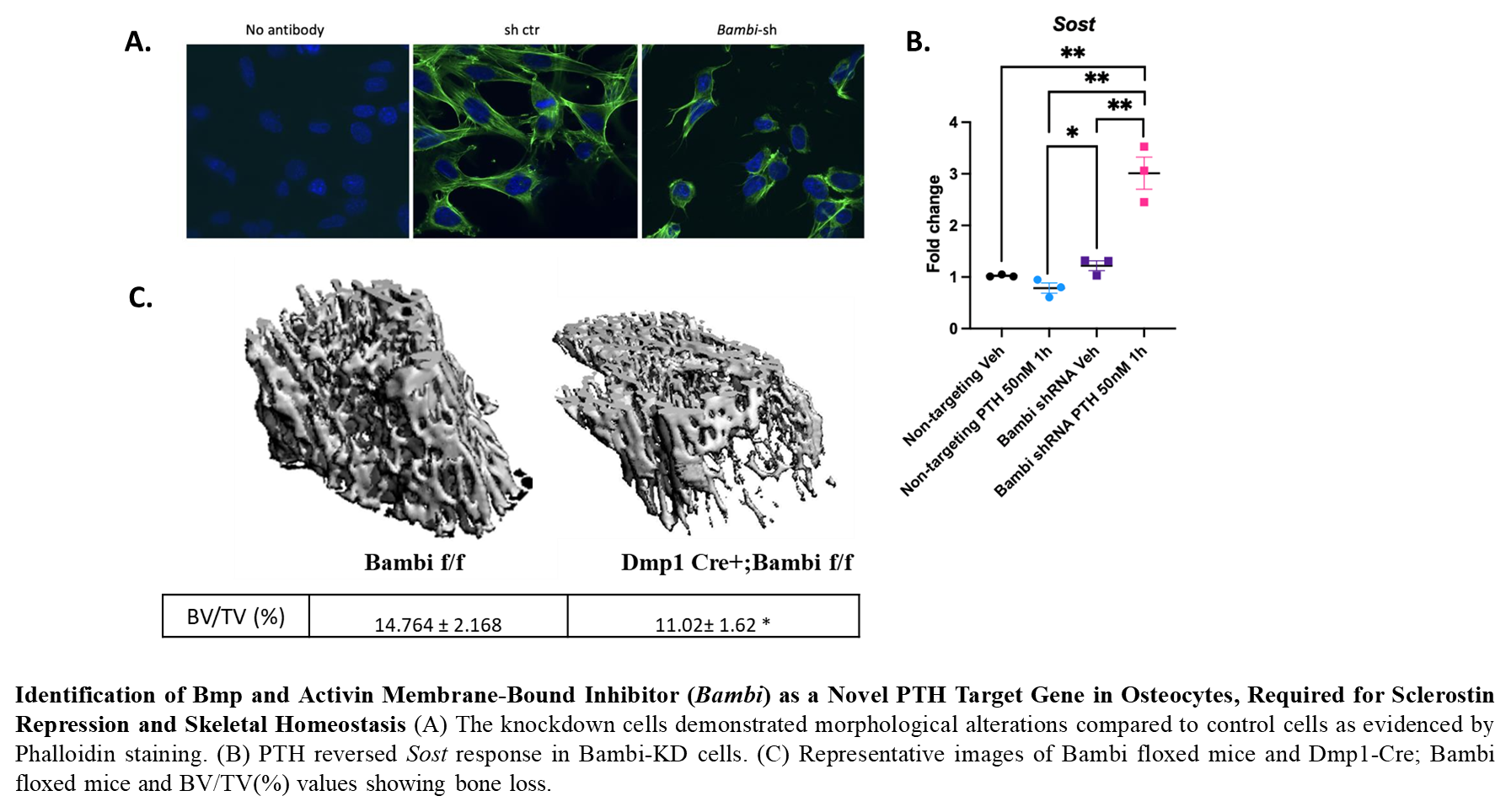
Pour en savoir + : 1123 - Identification of Bmp and Activin Membrane-Bound Inhibitor (Bambi) as a Novel PTH Target Gene in Osteocytes, Required for Sclerostin Repression and Skeletal Homeostasis. Kalpana Patel and al. ASBMR Meeting 2025.


